
A proposito di sindrome
S I N T E S I
Questo è ciò che la sindrome di Gorlin? DEFINIZIONE
DR Gorlin RJ, CHIRURGO-dentista e genetista, il suo corso
CARATTERE GENETICO sindrome del nevo basocellulare
A / GENERALE
B / GENE COINVOLTO NELLA basocellulare sindrome del nevo
1. Generalità
2. Teoria del doppio evento
3. trasduzione Way rattoppato / sonic hedgehog
Attori via di trasduzione del patch / di Sonic Hedgehog
La via di trasduzione del patch operativo / di Sonic Hedgehog
Segnalazione percorso rattoppato / Sonic hedgehog
Tipi di mutazioni e posizione
EVENTI GENERALI In dettaglio, il basocellulare sindrome del nevo
1 / EVENTI dermatologici
A / Naevi e carcinomi a cellule basali generale
B / palmo-plantare ipercheratosi Generale
C / eventi dermatologici
2 / SKELETON EVENTI
Calcificazioni A / cervello buste
B / forma del cranio anormale
Anomalie C / costali
D / anomalie vertebrali
E / osso anormale della mano e del piede
F / Altre manifestazioni scheletriche
3 EVENTI / OCCHI
A malformazioni congenite / del globo
B / Tumore processo globo
4 / Disturbi neurologici
A / Méduloblastomes Statistiche
B / Altre manifestazioni neurologiche
5 / ALTRI EVENTI GENERALI
A / ovariche fibromi
B / fibromi cardiaci
C / ipogonadismo negli uomini:
6 / EVENTI ODONTOSTOMATOLOGIQUES
A / I cheratocisti odontogene
B / Altri eventi odontostomatologiques
un / Inclusioni o malposizionamento dentale
b / prognatismo mandibolare
c / labbro e palato schisi
d / parestesia
e / La améloblastome
Questo è ciò che la sindrome di Gorlin? DEFINIZIONE
Sindrome di Gorlin, conosciuta anche come sindrome del nevo basocellulare (NBC) è una malattia ereditaria caratterizzata da una serie di anomalie dello sviluppo e di una predisposizione a vari tipi di cancro.
La prevalenza è stimata tra 1/57 e 1/256 000 000, con un rapporto maschi / femmine di 1: 1. Le manifestazioni cliniche comprendono la presenza di numerosi carcinomi a cellule basali (BCC), cheratocisti odontogene della mascella, ipercheratosi palmo-plantare, anomalie scheletriche, le calcificazioni ectopiche intracraniche e dismorfismi facciali (macrocefalia, schisi labiale palato e gravi anomalie oculari). Ritardo mentale si osserva nel 5% dei pazienti. Carcinomi a cellule basali (da papule aventi il colore della pelle a piastre ulcerative con un diametro compreso tra 1 e 10 mm) si trovano in genere sul viso, schiena e al torace. Il numero di carcinomi a cellule basali variava da pochi a diverse migliaia. Anomalie scheletriche (che influiscono sul profilo delle costole, vertebre e del cranio) sono frequenti. Può verificarsi anche occhio, genito-urinario e disturbi cardiovascolari. Tra i pazienti con sindrome di Gorlin, il 5-10% sviluppa il medulloblastoma, che è una probabile causa di morte precoce.
La sindrome è causata da mutazioni nel gene PTCH1 ed è trasmessa come autosomica dominante a penetranza completa ed espressività variabile. La diagnosi clinica si basa su criteri specifici. La diagnosi è confermata da mutazioni ricerca. La consulenza genetica è obbligatoria. La diagnosi prenatale è possibile con l'ecografia e analisi del DNA di cellule fetali ottenuti mediante amniocentesi o prelievo dei villi coriali. La principale diagnosi differenziale con la sindrome Bazex, trichoépithélium multipla e sindrome di Muir-Torre (si vedano questi termini). Il supporto richiede un approccio multidisciplinare.
Cheratocisti vengono rimossi dalla resezione chirurgica. In caso di carcinomi a cellule basali, la chirurgia è indicata quando il numero di lesioni è limitata. Altri trattamenti possibili sono ablazione laser, terapia fotodinamica e la chemioterapia locale.
La radioterapia deve essere evitata. Gli analoghi della vitamina A possono svolgere un ruolo preventivo contro lo sviluppo di nuovi CBC. L'aspettativa di vita non è significativamente alterata ma le complicanze può portare a una significativa morbidità. Un monitoraggio regolare da un team multidisciplinare (dermatologi, neurologi e odontoiatri) è essenziale. I pazienti devono evitare l'eccessiva esposizione ai raggi UV.
Editore (s) esperto (s)
DR Gorlin RJ, DENTISTA CHRIRURGIEN- e genetista, il suo corso

Robert James Gorlin è nato 11 gennaio 1923 a Hudson, New York, ed è morto di linfoma 29 agosto 2006 a Minneapolis, Minnesota, all'età di 83. Entrato nell'esercito durante la seconda guerra mondiale, ha poi studiato odontoiatria presso l'Università di Washington dove si è laureato nel 1947. Ha continuato la sua formazione ottenendo un Master di Chimica presso l'Università di Stato dello Iowa. Dopo numerosi incarichi accademici, nel 1956 ottenne un posto come professore e Direttore della Divisione di Patologia Orale presso l'Università del Minnesota, dove rimase fino alla morte.
La sua carriera ebbe una svolta nel 1940, quando Gorlin scopre un libro di Helen Curth dermatologo che fare con achantosis nigricans. Questa esperienza contribuirà a trasformare Gorlin in "syndromologiste". Il nigricans achantosis è una malattia della pelle caratterizzata da vegetare ipertrofia papillare e la pigmentazione localizzata principalmente nel ascelle, collo e le regioni genitali-femorale, dove la pelle appare ruvida, ispessita e squadrato. Questa condizione dermatologica ha forme maligne caratterizzate da una associazione con adenocarcinoma gastrico. Si tratta di una condizione dermatologica che colpisce altri sistemi del corpo. Questa scoperta ha conquistato Gorlin che poi chiesto se alcune malattie sistemiche possono presentare manifestazioni orali. Gorlin divenne noto a livello internazionale per il suo lavoro sulle manifestazioni orali di molte sindromi. Durante la sua carriera, Robert J. Gorlin ed i suoi colleghi hanno studiato e nominati circa 100 sindromi e partecipato l'isolamento e la caratterizzazione di geni responsabili di circa la metà di loro.
Il nome di Gorlin è diventata un nome comune, 7 familiare non solo nel campo della genetica, ma anche in medicina. Ciò ha indotto dermatologi, genetisti e patologi a considerare Gorlin come uno di loro. Robert J. Gorlin sarà uno scrittore molto prolifico. Ha scritto circa 600 articoli, 60 capitoli di libri e co-autore di 20 libri o modificato.Alcuni di loro sono stati anche tradotti in spagnolo, russo e giapponese. Il nome di Gorlin è diventata particolarmente inseparabile dalla sindrome del nevo basocellulare, anche comunemente nota come sindrome di Gorlin.
Nel 1960, descrive per la prima volta l'associazione di numerosi carcinomi a cellule basali cutanei compaiono su tutto il corpo comprese le aree non esposte al sole con malformazioni costali, lesioni ossee e difetti e tumori oculari .Egli considera queste anomalie come essere parti di un unico sindrome di quanto lo sia oggi con il termine sindrome di Gorlin. Esso aiuterà nella comprensione e la scoperta del gene-malattia, il gene PTCH.
Durante la sua carriera presso l'Università del Minnesota, una vasta conoscenza lo ha portato a tenere corsi di patologia, dermatologia, pediatria, ostetricia, ginecologia e otorhino gola. È stato anche Presidente della International Association for Dental Research, l'American Academy of Patologia Orale e la Società Internazionale di Biologia cranio-facciale. Per il suo grande contributo alla scienza, medicina e società, Robert J. Gorlin ha ricevuto molti premi prestigiosi in tutta la sua carriera. Robert J. Gorlin è quindi un dentista di formazione, che con la sua opera è stata in grado di attraversare i campi della medicina e della genetica, nonché Odontoiatria.
CARATTERE GENETICO sindrome del nevo basocellulare
A. GENERALI
Molto prima Gorlin RJ, questa entità clinica, conosciuta come il termine sindrome del nevo basocellulare, è già stato oggetto le descrizioni, sotto altri nomi. Nel 1894 Jarisch
descrive brevemente la combinazione di carcinoma a cellule basali con anomalie scheletriche in un paziente affetto da scoliosi. Non fa alcuna menzione di altri
caratteri della malattia.
Lo stesso anno, ha riferito Bianco storia familiare di un paziente con più carcinomi a cellule basali, che hanno continuato ad apparire in tutta la sua vita.
Nel 1932 Nomland descrive carcinomi basocellulari derivanti dalla congenita nevi pigmentati.
Nel 1939 Straith è il primo ad associare cisti mascellari a più carcinomi cutanei a cellule basali in tre membri di una famiglia.
Si attenderà fino al 1951 che i due dermatologi, Binkley e Johnson descrivono sia Tumori della pelle e cisti del mascellare e
cambiamenti del cervello.
E 'stato nel 1960 a Minneapolis come Gorlin, dentista e Goltz, dermatologo eseguire la prima descrizione completa di questa entità clinica che considerano una sindrome. Lo daranno il nome di sindrome del nevo basocellulare con più cisti della mascella e costole bifidi.
Da allora, la sindrome può essere designato con nomi diversi come la sindrome di Gorlin o Gorlin-Goltz Gorlin-Goltz facomatosi, sindrome del nevo basocellulare ...
I primi casi noti di sindrome del nevo basocellulare sono stati rivelati nel 1960 a causa di cisti dentigerous, costole bifidi e altre stimmate della sindrome di Gorlin sono stati trovati su due maschi adulti scheletri scheletri della collezione egizia l'Istituto di Antropologia dell'Università di Torino. Questi scheletri sono stati riesumati a Assyut nel Basso Egitto e risalgono al periodo dinastico. Periodo dinastico inizia con l'unificazione d'Egitto, una volta divisi in due regni, il Nord e il Sud, ed è intorno al 3000 aC.
La sindrome del nevo basocellulare è una malattia genetica ereditaria con autosomica dominante. Tuttavia, sono stati segnalati casi sporadici: nel 60% dei casi, nessun altro membro della famiglia è raggiunto e 35-50% di questi casi rappresentano néomutations. Pertanto l'assenza di una storia familiare non esclude la diagnosi di questa malattia. È causata da mutazione del gene PTCH, situato in 9q22.3-q31, che è un gene
soppressore del tumore.
Il basocellulare nevoide ha una penetranza elevata che è la frequenza con cui un gene manifesta i suoi effetti. Si è stimato al 97%. Ciò significa che il 97% del
individui con questo gene soffrono di almeno una delle manifestazioni della malattia.
La malattia ha anche una variabile espressività; espressività è la qualità del gene presente con termini variabili quantitativamente. La malattia si manifesta in tanti sintomi di una variabile ad un altro paziente. Ci sono più di quaranta criteri diagnostici. Tuttavia, ogni paziente con questa malattia non presentano tutti i criteri. I criteri diagnostici sono stati classificati in due categorie, maggiori e minori, a seconda
la loro frequenza di occorrenza. E 'possibile effettuare una diagnosi di cellule basali sindrome del nevo quando i pazienti presentano almeno due criteri maggiori o collegate
un criterio importante nella due criteri minori.
La prevalenza è 1/56000 con un rapporto sessuale maschile / femminile 3/1. Secondo Ghailan, la prevalenza è 1/60000 e gli uomini e le donne sono colpite in
stesse proporzioni. Questo raggiunto la parità tra i sessi è spiegato molto bene dalla sindrome autosomica dominante.
Secondo Wolff, la prevalenza varia da 1/60000 a 1/120000 e entrambi i sessi sono colpiti in ugual misura. Inoltre sindrome si verifica in una grande varietà di gruppi
Culturale e non sembra avere una predilezione per particolare tipo di pelle.
Sindrome del nevo basocellulare rimane una malattia rara come attualmente, solo 700 casi sono stati segnalati in letteratura. Tuttavia, il dentista è nella prima posizione per fare la diagnosi perché manifestazioni maxillofacciali dentali sono tra le prime manifestazioni di questa malattia.
B. GENE COINVOLTO NELLA basocellulare sindrome del nevo
1. Generalità
Il gene responsabile della sindrome del nevo basocellulare, il gene PTCH, è stato scoperto per la prima volta nel 1996 da due gruppi di ricerca. La sua scoperta è piuttosto recente
tutti i meccanismi genetici che portano alla malattia non sono ben compresi dalla comunità scientifica, alcuni elementi rimangono poco chiari.
Situato sul braccio lungo del cromosoma 9 e più precisamente in 9q22.3, questo gene è trovato mutato in pazienti con sindrome di Gorlin ma anche nei casi
sporadici carcinomi a cellule basali.
Questo gene svolge un ruolo importante nello sviluppo embrionale e il controllo della proliferazione cellulare.
In un articolo del 1982, Fitzpatrick fornisce una teoria molto diversa dei dati correnti di spiegare l'origine di questa sindrome. Gli scienziati del tempo le loro tecniche hanno, infatti, non è riuscito a rilevare l'anomalia genetica che conosciamo oggi. Ha detto che le anomalie multi-sistemici non hanno potuto spiegare l'esistenza di un difetto enzimatico. La presenza di tale difetto durante il primo trimestre di gravidanza potrebbe spiegare le anomalie scheletriche e calcificazioni avvolge il cervello.
Più tardi, una anomalia nel meccanismo enzimatico che controlla la pelle potrebbe portare alla formazione di nevi, carcinomi a cellule basali, pozzi palmo-plantare e cheratocisti odontogene. Tuttavia questa teoria non è sufficiente perché fa permetpas per capire l'origine di tumori neurologici come il medulloblastoma.
2. Teoria del doppio evento
I tumori di studio in sindrome del nevo basocellulare hanno mostrato l'esistenza di una perdita di eterozigosi per regione 9q, suggerendo che il gene PTCH potrebbe essere un gene soppressore del tumore. Questa nozione di anti-oncogene, il cui archetipo è il gene retinoblastoma (Rb), è basato sulla perdita di entrambi gli alleli di questo gene in un tumore (perdita di eterozigosi 13) dal modello proposto da Knudson nel 1971. Secondo la sua "teoria dei due eventi mutazionali ', la prima mutazione è responsabile della sindrome malformazioni e predispongono all'insorgenza di tumori e il secondo porterebbe alla comparsa di tumori.
Secondo Cohen, in modo che un gene soppressore del tumore è inattivato, sono richiesti due fenomeni. Il primo è il trasferimento di un allele. Il secondo fenomeno è la perdita dell'altro allele, nota come la perdita di eterozigosi (LOH o). Quando entrambi gli alleli sono persi, i tumori si verifica. Una perdita di eterozigosi è stata dimostrata per i carcinomi a cellule basali, cheratocisti odontogene e medulloblastoma, che sono tre delle principali caratteristiche sindrome di Gorlin.
Così, nella forme familiari sindrome di Gorlin, la prima mutazione può essere presente al momento del concepimento e si trova in tutte le cellule del corpo. Questo primo evento mutazionale, generando una predisposizione a sviluppare la sindrome del nevo basocellulare nella prole, viene trasmesso in una posizione dominante. Questo evento rimane latente e espone solo che se un secondo incidente si verifica nel cromosoma
omologa allo stesso locus, con conseguente comparsa sindrome di Gorlin. In casi familiari, l'individuo ha bisogno di un solo passo mutazionale (da quando ha ereditato la prima del suo materiale genetico). Con contro la comparsa sindrome di Gorlin in un individuo con una storia familiare
richiede il verificarsi di due eventi mutazionali successive nello stesso individuo.
3. trasduzione Way rattoppato / sonic hedgehog
Il gene PTCH è l'omologo umano di gene rattoppato in Drosophila. Questo gene è una segmentazione del gene Drosophila (o polarità) che si verificano nel percorso di
trasduzione patch / sonic hedgehog che è coinvolto nel controllo dello sviluppo embrionale e la proliferazione cellulare. Esso funziona come un regolatore del ciclo cellulare bloccando la divisione cellulare in assenza di ligando e attivando appena il collegamento è realizzato con il ligando.
Sindrome di Gorlin provoca danni al cervello, costole, vertebre, i membri ... rattoppato è espresso in questi tessuti.
Attori via di trasduzione del patchato hedgehog / sonic:
Anche se sono stati fatti grandi progressi negli ultimi anni per la comprensione del funzionamento del percorso di segnalazione patch / sonic hedgehog, molti
incertezze devono ancora essere chiariti in futuro.
- Il phenylthiocarbamide Patchato o proteina codificata dal gene PTCH è una glicoproteina composta da 1447 aminoacidi con domini transmembrana dodici,
due grandi anse extracellulari necessari per il legame del ligando SHH, un'ampia ciclo intracellulare, un dominio ammino-terminale intracellulare e un dominio intracellulare carbossi-terminale pure.
Si trova mutato in sindrome del nevo basocellulare, ma anche in carcinomi a cellule basali, e rari casi di medulloblastoma.
La maggior parte delle mutazioni sono mutazioni nonsenso che provocano la sintesi di una proteina tronca.(Lacombe) Gene rattoppato 1
- La proteina Sonic hedgehog (SHH), codificata dal gene localizzato in 7q36 Shh. Questa proteina 45kDa viene scissa in due domini: un dominio ammino-terminale di 20 kDa Zn avente un'attività idrolasi e un dominio carbossi-terminale di 25 kDa autocatalitico dotato
attività transferasi colesterolo.
Il ruolo del colesterolo in questo percorso di segnalazione non è completamente noto ma potrebbe svolgere un ruolo nel limitare la diffusione di SHH molecola e distribuzione
effetto spaziale.
- Infine, la proteina Smoothened (SMO), codificato dal gene localizzato nella 7q31-32 Smoothened.
È una proteina di 115 kDa, composta di 787 aminoacidi appartenenti alla famiglia dei recettori accoppiati alle proteine serpentina G (che mostra omologia alla famiglia recettori Wnt). Quest'ultimo ha un dominio extracellulare ammino-terminale, un dominio intracellulare carbossi-terminale, e due zone (terzo ciclo intracellulare e il dominio transmembrana settima) coinvolti nella segnalazione.
La via di trasduzione del patch operativo / di Sonic Hedgehog:
Patch è un recettore per SHH ruolo ligando. In assenza di SHH, SMO rattoppato e formare un complesso inattivo.Agisce rattoppato come
regolatore negativo, un inibitore di SMO.
Quando SHH viene legano al rattoppato, il secondo è quindi inattivato e SMO è libero di trasmettere il segnale all'interno della cellula.
Diversi meccanismi sono stati proposti per spiegare l'attivazione di SMO e trasduzione del segnale:
- In una prima ipotesi, rattoppato e Smo formano un complesso. SHH ligando al suo risultato Patchato recettoriale in un cambiamento conformazionale di quest'ultimo e quindi la dissociazione del complesso con patch / SMO. SMO sarebbe quindi liberi di trasmettere il segnale all'interno della cellula.
- Più di recente, una seconda ipotesi propone un regolamento non stechiometrico per rattoppato. Infatti, i test hanno dimostrato che il legame di SHH risultato diminutionde patchato e aumentate GOS nella membrana cellulare.Il rapporto tra
atched e SMO sarebbe non stechiometrica, vale a dire che regolano il rattoppato 16
Cambiamento SMO ma non costituiscono un vero e proprio complesso con esso (non ci sarebbe alcuna interazione fisica tra i due).
SMO è attivato e può eseguire la trasduzione del segnale al nucleo.
La precisa trasduzione del segnale via GOS non è ancora noto, tuttavia è noto che l'obiettivo finale è rappresentato dalla attivazione del fattore di trascrizione GLI (Cubitus interrotto omologo o CI Drosophila). Nelle cellule non esposta a SHH GLI tétramétrique formano un complesso con altre proteine, come Costal-2 (COS2) Fused (Fu) e soppressore del fattore fusa (SUFU). In questa forma, GLI può essere scissa in un frammento terminale 75 kDa amminico che può traslocare al nucleo e reprimere geni bersaglio in quest'ultimo.
In presenza di SHH ligando, i dissocia complessi e svolge tutto il suo attivatore ruolo GLI trascrizionale (accade nel nucleo e consente l'espressione di geni bersaglio).
I geni bersaglio sono:
- Wingless (Wg) membri di codifica della famiglia WNT nei vertebrati,
- Decapentaplegic (DPP) che codifica per la proteina morfogenetica dell'osso (BMP) e TFG beta
- Codifica PTC se stesso patch.
Questi geni sono essenziali per il normale sviluppo embrionale e la differenziazione di alcuni tessuti.
Il gene senza ali per una famiglia di glicoproteine coinvolte in processi di sviluppo quali la differenziazione, la migrazione, la proliferazione cellulare e la polarità. Espressione deregolamentato di questo gene è responsabile di anomalie dello sviluppo e oncogenesi.
Il gene decapentaplegic svolge un piccolo ruolo nella trasmissione delle informazioni alla differenziazione dorsoventrale del ectoderma embrionale.
Segnalazione percorso rattoppato / Sonic hedgehog
Ruolo nello sviluppo patchato:
un percorso di segnalazione Hedgehog gioca un ruolo importante nello sviluppo di cellule staminali in una varietà di tessuti. PTCH si esprime sia nella
lo sviluppo embrionale e negli adulti, suggerendo che esso continua ad avere un ruolo durante il periodo post-natale. Modelli sperimentali che esprimono impropriamente qualsiasi componente del percorso di segnalazione riccio rivelano significative anomalie dello sviluppo. La somiglianza tra queste anomalie e le caratteristiche della sindrome di Gorlin implica che l'eccessiva attivazione di questo percorso di segnalazione può essere la causa di anomalie dello sviluppo e dei tumori trovati in cellule basali lacnaevomatose.
Patch regola la trasmissione del segnale emesso dal SHH, segnale coinvolto in numerosi processi embryonaux come la formazione del tubo neurale ventrale, parti del cervello, arti o sviluppo dentale ...
Tipi di mutazioni e Posizione
Molte mutazioni sono state riportate dagli autori. Infatti, la sindrome del nevo basocellulare non può essere attribuito ad una singola mutazione. Secondo i pazienti, ci sono mutazioni nonsense, missenso, inversioni, delezioni ... che possono essere distribuiti su tutta la sequenza codificante del gene PTCH.
La maggior parte delle mutazioni portato alla creazione di un codone di stop che porta alla sintesi di una proteina tronca.
Queste mutazioni sono concentrate per lo più su ampia ansa intracellulare, l'ampia ansa extracellulare e la regione terminale amminico del gene PTCH.
Nessuna correlazione potesse essere stabilita tra la posizione della mutazione nel gene e il fenotipo osservato in diversi pazienti sindrome di Gorlin. Una caratteristica di questa sindrome è la mancanza di genotipo / fenotipo. Per gli individui con la stessa mutazione, i quadri clinici può essere molto variabile. Questo suggerisce la possibilità di l'influenza di altri fattori, come i fattori ambientali o genetici nello sviluppo della malattia.
EVENTI GENERALI In dettaglio, il basocellulare sindrome del nevo
1 / EVENTI dermatologici
A / Naevi e carcinomi basocellulari Generale:
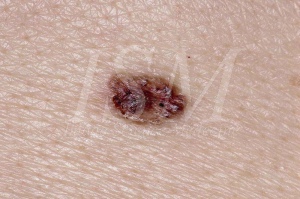
Nevo a cellule basali sono uno dei principali segni della sindrome del nevo delle cellule basali in modo tale che, in assenza di queste nevi; alcuni medici rimuovono emblema della diagnosi. Sono tra i criteri principali per la malattia, allo stesso modo che odontogeni cheratocisti. Hanno un grande potenziale evolutivo di degenerazione maligna in carcinomi a cellule basali.
Si trovano in oltre il 90% dei casi di sindrome di Gorlin. E 'stato stimato che 200 pazienti con carcinoma a cellule basali o più viene raggiunto sindrome di Gorlin. Più basale diagnosi carcinoma a cellule è fatta, quanto prima questa percentuale aumenta. Considerando che circa uno su cinque pazienti affetti da carcinoma basocellulare prima dell'età di 20 anni soffre di sindrome del nevo basocellulare.
Età di esordio:
Carcinomi basocellulari, come parte sindrome di Gorlin, compaiono in età più precoce rispetto alla popolazione normale. Essi possono essere presenti alla nascita o sviluppare durante l'infanzia. Più spesso, il loro verificarsi inizia durante la pubertà e quarant'anni, il 90% dei pazienti affetti da questa malattia hanno sviluppato almeno un carcinoma a cellule basali.
Numero: Il numero di carcinoma basocellulare è molto variabile. Sebbene casi clinici sono stati osservati con un singolo carcinoma, nella maggior parte dei casi, il numero è da venti a diverse migliaia.
Località:
La posizione di questi tumori può essere fatto sia sulle zone esposte al sole (e raggi ultravioletti quindi) rispetto alle aree non esposte. Le palpebre, naso, guance e sulla fronte sono i siti più colpiti, ma il collo, tronco e all'ascella sono spesso colpiti. Con contro, cuoio e arti sono, la maggior parte del tempo risparmiato. Aspetto clinico: nevo a cellule basali possono assumere aspetti clinici variabili.
Tre aspetti sono generalmente descritti:
- Aspetto evocando il contagium mollusco è una condizione della pelle contagiosa e inoculata in determinate condizioni. E 'causata da un virus ed è caratterizzata da piccole elevazioni di un bianco opaco o aumentato le dimensioni di una perla e la parte superiore è allargato da una depressione nel dell'ombelico.
- Aspetto di iperpigmentata grande nodulo.
- Aspetto evocando il carcinoma a cellule basali classica. Questa è la più comune. Nevo allora appare come una piccola superficie liscia del tumore traslucido. La colorazione è variabile, generalmente quella della pelle normale, a volte rosa pallido o grigio. La coerenza è molto più forte di quanto ci si aspetterebbe aspetto ma la base non è infiltrato. Le dimensioni variano tra 1 e 3 mm di diametro. Se uno qualsiasi di queste lesioni è aumentato in termini di dimensioni o inizia a sanguinare o ricoprirsi di una crosta, trasformazione maligna si sospetta. Carcinomi basocellulari che compaiono all'interno della sindrome del nevo basocellulare non possono essere chiaramente distinti da quelli che appaiono in un influenzato da questa sindrome paziente. L'unico elemento caratteristico sindrome di Gorlin è la loro presenza in gran numero e in tenera età e la loro posizione, che non è necessariamente in zone esposte al sole. L'aspetto clinico di questi tumori è altamente variabile da papule color carne marroni o placche ulcerosa. Essi possono essere scambiati per nevi o emangiomi. Le loro dimensioni variano tra 1 e 10 mm di diametro.
Istologia:
L'aspetto istologico del nevo basocellulare è quella di una proliferazione di cellule epiteliali intradermica, formando intervalli ben definiti, spesso con palisade periferico seduta. Lo stroma sottostante è ricco di fibroblasti. L'aspetto istologico dei pazienti carcinoma a cellule basali sindrome di Gorlin non è differente da quelli riscontrati in pazienti non sindromici. Quando nevi diventa il carcinoma basocellulare, le cellule si nota più grandi e più chiare, aumento del numero di mitosi, un carattere infiammatorio dell'infiltrato. Ha una stroma fibroso. La lesione può diventare papullaire o pedinato. Penetrazioni profonde, ulcere o invasioni possono verificarsi con infiltrazione linfocitaria. Essi possono essere pigmentati in massa o in periferia.
Influenza del sole:
Uno studio suggerisce che i pazienti con sindrome del nevo basocellulare presenterebbero una maggiore sensibilità ai raggi ultravioletti. Ciò è confermato dal fatto che solo il 14% dei pazienti con sindrome di Gorlin, nel nord-ovest dell'Inghilterra sviluppare carcinomi a cellule basali, contro il 47% in Australia. Sottolinea l'importanza della diagnosi prenatale (quando viene raggiunto un genitore) o la diagnosi quanto più precoce (per casi apparivano de novo) per limitare l'esposizione ai raggi UV al momento della diagnosi positivo.
Un altro studio, effettuato i rapporti che l'88% dei carcinomi a cellule basali nelle donne e 86% tra gli uomini nella popolazione generale si verifica in zone del corpo esposte al sole contro il 59% tra le donne e il 65% tra i maschi portatori della sindrome del nevo basocellulare . Ha concluso che la distribuzione anatomica delle lesioni suggerisce che il sole non è un fattore essenziale per lo sviluppo di carcinomi a cellule basali. La loro presenza in numero maggiore nelle aree esposte, tuttavia, suggerisce che il sole può esacerbare lo sviluppo di carcinomi a cellule basali e deve proteggersi. Egli non ha riportato alcuna correlazione significativa tra il numero di carcinomi a cellule basali e il numero totale di ore di esposizione alla luce solare.
Solo circa il 40% degli afro-americani con la sindrome di Gorlin sviluppare carcinomi basocellulari contro il 90% dei caucasici. Lo studio in 105 pazienti con sindrome di Gorlin mostra che solo il 38% degli afro-americani hanno una o più carcinomi a cellule basali contro il 97% dei caucasici. Inoltre, caucasici sviluppare carcinomi a cellule basali più presto. Infatti, circa il 50% dei caucasici sviluppare il loro primo carcinoma 21,5 anni e il 90% a 35 anni contro il 20% e il 40% a parità di età in afro-americani
Il più piccolo di frequenza carcinomi a cellule basali nel gruppo di afro-americani possono essere dovuti a una migliore protezione contro i raggi ultravioletti a causa della pigmentazione della loro pelle.
Influenza delle radiazioni ionizzanti:
Le radiazioni ionizzanti è stata anche implicata nello sviluppo di questi tumori. Infatti, i pazienti sottoposti a trattamenti di radioterapia hanno spesso numerosi carcinomi a cellule basali nelle zone irradiate. Essi appaiono in media oscillante in un periodo compreso tra 6 mesi e 3 anni dopo l'irradiazione contro una media di 21 anni con un non paziente con sindrome di Gorlin.
Va notato che uno dei primi segni della malattia è medulloblastoma. Ora è generalmente trattato con radioterapia. Il rischio di sviluppare BCC nella zona irradiata è fortemente aumentata. Inoltre, questi carcinomi sono molto più aggressive e quindi più difficile da trattare. Per queste ragioni, dobbiamo minimizzare i trattamenti di radioterapia e incoraggiare trattamento chirurgico.
Evolution:
Carcinomi a cellule basali appaiono come parte sindrome di Gorlin sembrano essere più aggressivo, più difficile da trattare. Si può arrivare fino a provocare la morte del paziente per invasione (metastasi nel cervello e nei polmoni principalmente) o resistenza al trattamento. Trasformazione maligna si verifica soprattutto dopo la pubertà. I tumori più aggressivi più spesso colpiscono le palpebre e il naso e causare danni ai tessuti che potrebbero portare a una sfregio significativo di pazienti.
B / palmo-plantare ipercheratosi Generale:
Hanno anche incontrato sotto i termini della "fosse", fori o pozzi palmoplantar. 26 Frequenza: Che si verificano in tenera età, la loro presenza può essere un ausilio importante per la diagnosi precoce della sindrome del nevo basocellulare perché sono presenti in circa il 65% al 80% dei casi. Si sono incontrati con circa la stessa frequenza in tutti i gruppi di età. Palmar ipercheratosi mani dopo immersione in acqua per alcuni minuti.
Aspetto clinico:
Essi corrispondono a piccoli fori che sono difetti puntiformi nello strato corneo, che è parzialmente o totalmente assente. Raramente sopra 1 o 2 millimetri di diametro e 1 mm di profondità, il fondo può essere rosa o, se lo sporco accumulato all'interno, scuro. Queste lesioni si incontrano solo sulle mani e sui piedi e sono asintomatici. Quando la diagnosi clinica è difficile, il praticante può immergere per qualche minuto le mani e / o piedi del paziente in acqua tiepida. Le fosse vengono visualizzate in modo più chiaro allora.
Istologia:
L'esame istologico di un box palmari ha mostrato un piccolo anteriori cellule basali proliferative. Anche se c'è una disorganizzazione cellulare entro questo fronte, una fessura tra la parte anteriore e lo stroma adiacente non è stata osservata. La proliferazione vascolare mite e la fibrosi sono stati osservati nel derma adiacenti. istologique
In conclusione, fosse palmoplantar sono una caratteristica manifestazione sindrome di Gorlin. Essi sono un aiuto importante per la diagnosi di questa sindrome a causa della loro elevata frequenza e età del loro verificarsi. Per queste ragioni, sono tra i principali criteri diagnostici per la malattia, con carcinomi a cellule basali e cheratocisti odontogene.
La frequenza delle cisti della pelle in relazione alla sindrome del nevo basocellulare è tra il 35 e il 75% a seconda dello studio. Le cisti epidermoidi di solito si verificano sulle aree dei pilleuses corpo e raramente in zone non pilleuses come le palme e le piante dei piedi. Questa posizione tipica alle estremità del corpo sarebbe una manifestazione clinica distintivo di sindrome del nevo basocellulare.
Questa descrizione istologica è molto simile a quello delle cheratocisti odontogene, uno dei principali criteri diagnostici della sindrome del nevo basocellulare. Questi pelle cheratocisti istologicamente parenti delle cheratocisti dei mascellari, può essere un marchio tipico della sindrome del nevo basocellulare.
In conclusione, la scoperta di cisti cutanee situati principalmente sulle mani e sui piedi, alcuni hanno una forte somiglianza con istologicamente cheratocisti odontogene, deve essere considerato un "perle clinici" che ci possono condurre verso una diagnosi di sindrome di Gorlin .
c / Altre manifestazioni dermatologiche
Altre manifestazioni dermatologiche trovano riportato in letteratura, ma a causa della loro bassa frequenza di occorrenza, non sono chiaramente descritti. I pazienti possono così avere lipomi, milia di grano, macchie "latte" o pendoli di mollusco. Lipoma posta sopra la scapola destra. Da notare anche la presenza di numerosi nevi cutanei. I Pendoli Mollusca sono tumori fibrosi e la pelle flaccida. Essi possono essere planare e placcato, la sensazione depresso, o, al contrario, le proiezioni e anche peduncolato. Il milia è una rara malattia della pelle caratterizzata dalla formazione di piccole elevazioni traslucido luminoso, le dimensioni di uno spillo prodotta da degenerazione colloide (trasformazione di cellule in una sorta di gelatina) degli strati superiori il derma.
2 / SKELETON EVENTI
Calcificazioni A / cervello buste
È possibile incontrare in bambini fisiologiche o patologiche calcificazione intracranica. I primi sono molto rari. Questi ultimi sono più comuni e sono generalmente descritti in associazione con tumori (come medulloblastoma), sindromi neuro-cutanea, infezioni e malattie neurodegenerative. Nella popolazione generale, calcificazioni intracraniche appaiono in media circa 12 anni, mentre nei pazienti con sindrome di Gorlin, sono visibili dai primi anni di vita. Essi possono quindi essere utilizzati come un primo indicatore per la diagnosi della sindrome del nevo basocellulare. Di solito si riferiscono a falso cervello, che è parte della dura madre formando una partizione centrale tra due emisferi.La loro frequenza nei pazienti sindrome di Gorlin dimostra molto importante, dal momento che oscilla tra i 65 e l'85% secondo gli autori. Essi rappresentano il segno radiologico più frequente. Calcificazioni del tentorio si riscontrano anche, ma molto più raramente sul cervello sbagliato. La tenda del cervelletto è la parte della dura madre tra il cervello e il cervelletto. È anche possibile incontrare con agenesia del corpo calloso, ma questa anomalia rimane raro.
B / forma del cranio anormale
I pazienti con sindrome del nevo basocellulare presentano anomalie della forma del cranio. Queste anomalie, pur appartenendo a criteri minori della malattia, la diagnosi di grande interesse in quanto sono visibili dalla nascita. In alcuni pazienti il cranio è anormalmente grande ribalta con bozze frontali e parietali-temporale. Le sopracciglia sono prominenti. Il ipertelorismo (eccessiva distanza degli occhi) è comune e prognatismo è talvolta segnalato. La base del naso è crollato e ampliato. Una deviazione del setto nasale può anche essere osservato Uno studio su 105 pazienti con sindrome Gorlin, macrocephaly si osserva con maggior frequenza (50% dei casi) in questi pazienti rispetto alla popolazione normale. È lo stesso per hypertélorisme che è presente nel 42% dei casi.
Anomalie C / costali
Anche se appartiene ai criteri minori della malattia, anomalie costali sono presenti in circa il 42% dei pazienti, mentre la loro frequenza nella popolazione è molto bassa. Essi sono in gran parte presenti e sono principalmente rappresentati da: - bifidités - di agenesia - sinostosi costale. La sinostosi è una saldatura totale di due ossa adiacenti.È anche possibile osservare anormale allargamento anteriore o posteriore estremità delle costole. La terza, quarta e quinta costola sono più comunemente colpite da questi difetti, anche se tutte le costole si possono ottenere. Alcuni autori come Kimonis deciso di classificare le anomalie costali tra i principali criteri della malattia per due ragioni. La prima è la loro alta frequenza (42%) dei pazienti rispetto alla popolazione generale. La seconda è che queste anomalie possono essere facilmente individuati al momento della nascita e rappresentano quindi un importante elemento diagnostico della sindrome del nevo delle cellule basali nella popolazione pediatrica. Applicando i criteri di Kimonis, una diagnosi precoce della malattia può essere eseguita in un gran numero di pazienti. Infatti, altri criteri importanti per la malattia, come cheratocisti odontogene o più carcinomi a cellule basali non sono presenti alla nascita; Essi appaiono per lo più durante l'infanzia o l'adolescenza. I medici dovrebbero essere molto attenti ai minori segni clinici della malattia durante l'infanzia. La presenza della sindrome di Gorlin deve essere ricercata nei bambini nati con anomalie costali, fibroma cardiaco, palato o labbro leporino, polidattilia o macrocefalia.
D / anomalie vertebrali
Secondo uno studio di 105 pazienti con sindrome di Gorlin, i casi di scoliosi sono stati più frequentemente rilevati nel modo in cui il gruppo di persone colpite. Tuttavia, questa differenza non dimostra grande abbastanza per essere significativo. Individui malati sembrano avere scoliosi più gravi rispetto alla popolazione generale e il più delle volte si trova sui siti di anomalie dello sviluppo, come fusioni emi-vertebre o corpi vertebrali. Queste anomalie dello sviluppo si riscontrano nel 31% dei casi. Casi di spina bifida sono stati segnalati, ma con una frequenza leggermente diversa rispetto alla popolazione generale. La spina bifida è un difetto che consiste in una fessura dell'asse saldatura posteriori dei punti ossificazione colonna vertebrale predefinita su una o più vertebre, attraverso le quali sono ernia nella forma di una più o meno grande tumore, la meningi e il midollo volte con una quantità variabile di liquido cerebrospinale. Colonne cervicali e toraciche sono i più colpiti da questo difetto nei pazienti sindrome di Gorlin
E / osso anormale della mano e del piede
Polidattilia non è una deformità molto comune in quanto si verifica solo nel 3% dei casi, tuttavia, è facilmente rilevabile e presenti dalla nascita. La sua diagnosi è facile. Sindrome pazienti Gorlin può anche avere un accorciamento del quarto e / o quinto metacarpo. Queste malformazioni sono tra i criteri minori della malattia perché sono infrequenti.
F / Altre manifestazioni scheletriche
Sprengel deformità: 11% delle persone con sindrome del nevo basocellulare ha una deformità congenita Sprengel o elevazione della scapola. Questa anomalia prevede lo spostamento in alto e verso l'interno di una o entrambe scapola con deformazione e talvolta attaccamento al dorso dell'osso spostato. Tale incremento è dovuto ad un guasto della normale posizione di discesa dell'osso durante la vita fetale.
3 EVENTI / OCCHI
La frequenza di coinvolgimento oculare rimane poco chiaro. Questo è perché occupano poco spazio nella descrizione sindrome di Gorlin, come studi più pubblicati su questa malattia trattano criteri principali. Anomalie oculari che non sono oggetto di una menzione, non una vera descrizione.
A malformazioni congenite / del globo
I principali difetti dell'occhio riscontrati sono la cataratta, il glaucoma e coloboma cui frequenze combinate sono stimati a meno del 14% entro il Gorlin.
Cataratta è una malattia dell'occhio che porta alla opacità del cristallino o di sua capsula.
Il glaucoma è una patologia oculare caratterizzata da un aumento della pressione oculare superiore a 20 mm Hg. È dovuta al disagio nel normale flusso di umore acqueo.
Coloboma è un difetto della lente costituita da un intaglio periferico, singolo o multiplo.
Il ipertelorismo o eccessiva distanza tra gli occhi, è anche spesso soddisfatte in quanto fa parte delle caratteristiche tipiche del viso del sindrome del nevo basocellulare.
In aggiunta a queste anomalie, è anche possibile trovare casi di strabismo, cataratta, microftalmia o retinite pigmentosa. La retinite pigmentosa è un processo degenerativo della retina, bilaterali, famiglia ed ereditaria. Appare durante l'infanzia ed è caratterizzata da una progressiva perdita di acuità visiva e del campo visivo, portando alla fine alla cecità
B / Tumore processo globo
Le palpebre sono spesso sede di nevi a cellule basali a rischio di trasformazione maligna. Questa trasformazione può essere manifestata con l'istituzione di una ulcerazione torpida (vale a dire, che non si evolve e non regredisce) e invasivi o da infiltrazione in profondità con coperchio mutilazioni. Carcinomi a cellule basali delle palpebre sono generalmente più aggressivi e più difficile da trattare. Inoltre, il loro trattamento può causare una mutilazione facciale significativo per il paziente.
Calazio sono stati anche segnalati caso. Calazio è un piccolo tumore palpebra da infiammazione cronica di una ghiandola di Meibomio, ghiandole sebacee nella cartilagine tarsale, che costituirà il quadro delle palpebre.
4 / Disturbi neurologici
A / Méduloblastomes Statistiche:
Nella popolazione generale, medulloblastomi sono i tumori cerebrali più frequenti nell'infanzia e colpiscono circa il 20% di tutti questi tumori. L'età media della diagnosi è di 6 anni e 3 mesi ed i ragazzi sono colpiti in misura maggiore rispetto alle ragazze. Il trattamento tradizionale di questi tumori è costituito da escissione chirurgica in combinazione con la radioterapia e la chemioterapia. Il tasso di sopravvivenza a 5 anni è compresa tra il 60 e l'80%.Circa il 5% inferiore a 5 anni dei pazienti con medulloblastoma, vengono raggiunti da sindrome di Gorlin. Questo tasso scende a 1 o 2% nel caso degli adulti. L'età media di diagnosi medulloblastoma nei pazienti è molto prima, perché è di 2,1 anni.
Per questo motivo, è molto importante pensare alla presenza del medulloblastoma nei primi anni di vita dei bambini a rischio di sviluppare la sindrome del nevo basocellulare.
La sindrome pazienti hanno un tasso di sopravvivenza più rispetto ai non-pazienti.
Trattamento:
La resezione chirurgica, integrato da radioterapia e / o chemioterapia è generalmente proposto per sradicare medulloblastomi. In caso di sindrome di Gorlin, l'età di insorgenza di questi tumori è precedente; La terapia d'oro più di radiazioni è praticato, prima il rischio di sequele a lungo termine (come i disturbi della crescita o disfunzioni endocrine) è importante. Inoltre, le aree irradiate, il rischio di sviluppare BCC o meningiomi è notevolmente aumentata. Questi carcinomi a cellule basali risultano essere molto più aggressivi e resistenti al trattamento.
Per queste ragioni, il trattamento con radioterapia in persone infettate sindrome deve essere evitata quando possibile.
Medulloblastomi soddisfano anche i trattamenti di chemioterapia 42, questa modalità di trattamento dovrebbe essere favorito, almeno tra i pazienti più giovani.
Alcune funzioni sindrome di Gorlin, presente in tenera età, come ritardo mentale o calcificazioni della falce, può essere interpretato solo come manifestazioni causati dal tumore. Inoltre, i più classici segni della malattia, come lesioni cutanee o cheratocisti mascellari, appaiono solo successivamente.
Per questi motivi, la sindrome del nevo basocellulare può essere difficile identificare a questa età, nonostante la presenza del tumore.
Alcuni autori ritengono che il medulloblastoma dovrebbe diventare uno dei principali criteri della malattia, perché è parte, con le alterazioni ossee, i primi segni. Indagini genetiche e gli esami clinici rigorosi devono essere offerte, alla ricerca di una possibile sindrome di Gorlin, tutti i pazienti con medulloblastoma prima di 3 anni e le loro famiglie.
B / Altre manifestazioni neurologiche
Meningiomi:
Solo 10 casi di meningioma naevomatoses basali associate sono stati riportati in letteratura. L'età dei pazienti è tra i 18 ei 64 anni. Non c'è predominanza sessuale. Questi tumori sembrano crescere in zone radiazioni indotte dal trattamento di un medulloblastoma precedente.
Ritardo mentale:
I casi di ritardo e di apprendimento difficoltà mentali sono stati riportati anche in circa il 5% dei pazienti.
5 / ALTRI EVENTI GENERALI
A / fibromi ovarici:
Le ragazze e le donne affette da sindrome di Gorlin possono presentare fibromi ovarici. Il fibroma è un tumore benigno del ovarica più comune e rappresenta circa il 20% di tutti i tumori ovarici. Nella loro relazione iniziale nel 1964, Gorlin Goltz e hanno stimato che i fibromi ovarici erano presenti nel 75% delle donne affette da sindrome di Gorlin.
Una serie di studi condotti di recente ha mostrato una prevalenza compresa tra il 14 e il 24%. Età di media portata è 30,6 anni, ma può variare da 16-45 anni. Si noti, tuttavia, che alcuni di questi tumori sono stati trovati in pazienti di età inferiore 3,5 anni. Per questo motivo, la sindrome pazienti devono essere sottoposti a loro prima ecografia prima dell'età di 13 anni e di avere regolari cure ginecologiche. Fibromi ovarici crescono lentamente e una dimensione media di 6 cm di diametro, anche se alcuni possono andare fino a 30 cm di diametro.
E 'stato riportato che i fibromi ovarici incontrate nella sindrome di Gorlin sono più spesso bilaterali e calcificata che nella popolazione generale. Nella maggior parte dei casi, questi tumori sono completamente asintomatici. Essi non interferiscono con la fertilità e pazienti raramente sottoposti a trasformazione maligna. Il trattamento di questi tumori è chirurgica.
I fibromi B / cuore:
I casi di fibromi cardiaci sono stati segnalati in relazione alla sindrome del nevo basocellulare, ma sono rari. Sono 3 al 5% di tutti i pazienti con un fibroma cardiaca. Essi rappresentano solo il 5% delle neoplasie cardiache. Essi appaiono durante l'infanzia e nel 85% dei casi nei bambini sotto i 10 anni. Il loro posto preferito è il setto interventricolare. Questi tumori si trovano in fretta perché appaiono molto presto nella vita e può portare alla morte precoce di un paziente.
C / ipogonadismo negli uomini:
Gli uomini con di sindrome di Gorlin possono presentare molto raramente ipogonadismo. Ipogonadismo è la condizione di un soggetto la cui gonadi avere insufficiente secrezione interna.
6 / EVENTI ODONTOSTOMATOLOGIQUES
A. cheratocisti odontogene
1. Generalità
Descritta per la prima volta da Philipsen nel 1956, è un cheratocisti odontogena, secondo l'Organizzazione mondiale della sanità o OMS, "un benigno uni- o multi-celled, intraossea origine odontogena, con uno strato di caratteristiche stratificato parakératinisé squamose epitelio e il potenziale di comportamento aggressivo e infiltrante ". OMS raccomanda anche il termine "tumore", come questo meglio riflette la natura di neoplastica (comportamento aggressivo, grande espansione, la tendenza a recidivare ...). La cisti primordiale termine è stato a lungo utilizzato come sinonimo del termine cheratocisti, mantenendo confusione. In effetti, tutte le cisti primordiali non sono cheratocisti in quanto non tutti cheratinizzazione. Al contrario, tutti cheratocisti non sono cisti essenziali nella misura in cui non si sviluppano sempre al posto di un dente.
La cisti epidermoide termine è, nel frattempo, un sinonimo. Essi rappresentano circa l'11% di tutte le cisti dei mascellari. Essi terzo posto, dopo il radicolare e cisti follicolari. Essi appaiono tre volte più frequentemente nella mandibola che nella mascella.
Le cheratocisti odontogene si verificano in tutte le età, anche se una frequenza cumulativa si osserva tra i dieci ei 40 anni nella popolazione normale. Per contro, nei pazienti sindrome di Gorlin, la frequenza di picco è precedente ed è quindi nella prima decade di vita. Queste cisti costituiscono dunque un sintomo precoce di malattia. Ciò conferma il luogo della scelta del dentista nella diagnosi precoce della sindrome del nevo basocellulare. Essi sono il più delle volte scoperto per caso durante una radiografia standard. La percentuale di pazienti asintomatici varia a seconda dello studio 34-50%. Inoltre possono essere rivelati dalla comparsa di sintomi come gonfiore, dolore più spesso; ma trisma, cellulite, un'eruzione ritardo o malposizionamento dentale tra gli altri ...
Si noti che tutti questi sintomi non sono specifici di cheratocisti odontogene. La presenza di cheratocisti odontogene in un giovane paziente o la comparsa di ricorrenti o più cheratocisti devono allertare il medico e verso una possibile diagnosi di sindrome di Gorlin. Circa il 5% delle cheratocisti sono associati a questa sindrome. Si trovano nel 80% dei pazienti con sindrome Gorlin quando sono presenti solo 5-7% della popolazione generale. Ci sono diverse teorie per spiegare l'eziologia di cheratocisti odontogene.
Alcuni cheratocisti odontogene svilupperebbero dalle piastre dentali.
Infatti, in seguito alla formazione di germi dentali, strisce dentali dissolvono in entrambe le mascelle. Durante questo fenomeno, isolotti e gruppi di cellule epiteliali possono persistere nel tessuto connettivo. Sarebbe epiteliale di questi residui, in seguito all'attivazione da ancora incognite sono la causa della formazione di cheratocisti. Secondo altre, che fuoriescono dal reticolo stellate dell'organo smalto o strato basale dell'epitelio della mucosa orale. Infatti, tale altra teoria presuppone che le ramificazioni dello strato basale sono la causa della formazione di cheratocisti. Un argomento a sostegno di questa teoria è che le recidive osservati di queste cisti, anche quando la resezione chirurgica dei segmenti ossei interessati. Questo presuppone che l'origine della ricorrenza è al di fuori di questo osso e probabilmente nel tessuto molle adiacente. Va notato che queste due teorie non sono incompatibili in quanto sia la lamina dentale come mucosa orale sono responsabili tessuti ectodermiche.
2. Caratteristiche
La diagnosi istologica di cheratocisti odontogene può essere fatto con l'analisi histolopathologique di esso. Esame radiologico può certamente guidare il professionista, ma in nessun caso fare una diagnosi definitiva. L'esame consiste nella analisi dei contenuti e delle pareti cistica si ottiene dopo resezione completa della cisti o dopo l'assunzione di un frammento di esso durante il suo drenaggio marsupializzazione. Le principali caratteristiche istologiche elencate dalla maggior parte degli scrittori sono quelli di Pinborg e Hansen nel 1966:
- Fodera epitelio squamoso solito molto fine ed uniforme di spessore, con i germogli poco o nessun gusto e con una forte attività mitotico;
- Uno strato di cellule basali ben definito. Le cellule che compongono sono forme cubiche o cilindriche e disposti più spesso in una disposizione a palizzata;
- Un sottile strato, da quattro a otto strati cellulari di cellule spinose spesso presentano un edema intracellulare;
- Cheratinizzazione è, di solito, paracheratosica (presenza di nuclei). Essa si verifica in 83-97% delle cheratocisti. A volte è possibile incontrare una specie di cheratinizzazione orthokeratosic (senza nucleo) o entrambi para e orthokeratosic. Per alcuni autori come Martin, il tipo orthokeratosic deve essere individualizzato perché sarebbe meno aggressivo e meno ricorrente come una specie paracheratosica;
- Strato di cheratina è spesso ondulato. L'unica cheratinizzazione non è uno specifico criterio di cheratocisti perché altri le cisti odontogene in grado di produrre la cheratina.
- La parete della cisti fibrosa è solito sottile e non fa di solito l'infiammazione;
- Un congiuntiva sottile guscio spesso contenenti isole o cisti epiteliali "ragazze" isolato periferici. Il contenuto del lume della cisti ha un aspetto liquido, pallido o giallo crema, più bianco, più spessa. C'è una presenza di cheratina, ma in quantità variabili. In caso di reazione infiammatoria si trova in questa cristalli contenuto di colesterolo (essendo in forma di fiocchi lucidi), corpo ialino, polinucleari e ciuffi batteriche. Inoltre, quando la parete cistica è infiammato, l'epitelio è ispessita e gradualmente si trasforma in un epitelio cheratinizzato non abbastanza simile a quello odontogeni cisti infiammatorie banali. FNA è anche un importante test per la diagnosi di cheratocisti odontogenic.
, Infatti, la presenza di questo cheratina FNA è un elemento di aiuto diagnostico (ma che da sola non è sufficiente per fare una diagnosi definitiva perché altri cisti come cisti dentigerous, radice primaria o hanno un epitelio superficie che può essere cheratinizzato).
Tuttavia, come sopra descritto, la presenza di reazione infiammatoria indotta progressiva perdita di parakératinisation di cheratocisti e quindi talvolta possono impedire una corretta diagnosi citologica. Uno degli altri elementi di FNAB di rinvio ad uno cheratocisti diagnostica è la stima del contenuto proteico della cisti.
Quando questo contenuto viene misurata almeno 4 grammi di proteine per 100 ml di liquido, il cheratocisti diagnostico è quindi relativamente sicuro. Per questa misurazione, la presenza di infiammazione ha anche una cattiva influenza.
Infatti, tende ad aumentare il contenuto proteico e quindi partecipato alla creazione di falsi negativi. Ci sono anche metodi per realizzare la diagnosi prima di rivolgersi a un intervento chirurgico. Queste tecniche non sono ancora pratica comune. La prima è la ricerca di KAC antigene (antigene cheratocisti) nel fluido cisti. Questa presenza conferma la diagnosi. La seconda è la determinazione dell'espressione immunoistochimica di citocheratina 10.
Infatti, è un indicatore delle epiteli cheratinizzate. Si trova in circa il 50% delle cheratocisti odontogene, ma anche nel 10% delle cisti radicolari e 3% di cisti dentigerous. Tuttavia lo studio dell'espressione della citocheratina questo non sarebbe in cheratocisti penetranti come queste lesioni sembra perdere l'espressione dopo la decompressione.
3. Le caratteristiche cliniche e radiologiche
Località:
La sede odontogenico cheratocisti più spesso nella mandibola nella mascella. Infatti, si verifica tre volte più spesso nella mandibola, soprattutto a livello angolazione e il ramo, seguita da punti della superficie di primi e secondi molari e sedi più anteriore.
Presso la sede della cheratocisti mascellare è un po 'prima nella regione prémolomolaire e il canino ed eccezionalmente la regione anteriore.
Età di esordio: Le cheratocisti odontogene associate alla sindrome di Gorlin sviluppa, in generale, durante la prima decade di vita e raggiungono il loro picco di incidenza tra i 20 ei 30 anni, a differenza di non associata a questa sindrome e cheratocisti vengono scoperti più tardi, durante il quarto decennio (età media 37 anni).
A causa di questo primo sviluppo, costituiscono uno dei primi sintomi evidenti della malattia e confermano il luogo di scelta per l'odontoiatria nella diagnosi di questa sindrome.
Dimensione:
Il loro volume è più variabile, da pochi millimetri a un allagamento di un emi-mandibola (18). Poiché essi sono spesso scoperti dopo, durante un esame radiologico di routine, non è raro che hanno già raggiunto un notevole volume. La loro lunghezza media di 5 centimetri. (34)
Rapporto tra i sessi:
Nelle normali uomini di popolazione sono colpiti più spesso delle donne con un rapporto di sesso di 2/1. Con contro nei pazienti con sindrome del nevo basocellulare, la differenza tra i sessi non esiste e addirittura sembra che le donne sono colpite leggermente più spesso rispetto agli uomini. (5) Sintomi: La maggior parte delle cheratocisti odontogene è privo di qualsiasi sintomo. In effetti, il 34 al 50% sono asintomatici e quindi scoperto solo durante una visita odontoiatrica di routine o radiologico. Quando sono presenti sintomi, si verificano più spesso dalla comparsa di un arco o gonfiore osso progressiva può essere dolorosa.
I fenomeni infiammatori non sono poi infrequenti e sono caratterizzate dalla presenza di ascessi, cellulite o fistulisations addirittura a volte accompagnati un Lockjaw. Al di fuori di questi fenomeni, il cheratocisti può essere scoperto a causa di disallineamenti, malocclusioni dentali o ritardi, può causare eruzioni cutanee, e che si sfideranno il praticante.
Va notato che tutte queste manifestazioni non sono affatto specifici cheratocisti odontogene e richiederanno un esame radiologico e soprattutto istopatologico fine di confermare la diagnosi.
Crescita:
Una delle principali caratteristiche cliniche di cheratocisti odontogene è il loro forte potenziale di crescita. Questo è anche uno dei criteri che hanno portato l'OMS a considerarli come neoplasie benigne. Varie teorie sono state avanzate per spiegare questa caratteristica:
- Una prima spiegazione potrebbe essere l'esistenza di un enzima collagenasi all'interno cheratocisti. Sarebbe responsabile di un'attività collagenolitica che spiega questo forte potenziale di crescita;
- Un'altra teoria è l'esistenza di riassorbimento osseo indotto da prostaglandina E e F secreto dalla cisti;
- Infine, una maggiore osmolarità del liquido cistico e una maggiore potenza mitotico alla parete cistica potrebbe anche essere responsabile di questa forte crescita. Estendere queste cisti è all'interno dell'osso sostituendo spongiosa piuttosto che si estende lateralmente come altre cisti verso le regioni corticali e periostali.
Quando cheratocisti sono ingombranti, sono punzonate sui bordi delle ossa e dei denti, corticale 53 (solitamente esterno) viene quindi assottigliato, a volte forata. I casi di fratture mandibolari deformità facciali e / o maggiori sono stati segnalati, ma sono comunque molto rari. Gli elementi nobili come il fascio vascolo-nervoso del canale alveolare inferiore sono, a loro volta, sempre rispettati, ma possono essere respinti (e molto raramente causare parestesia).
Ricorrenza, meccanismo di recidiva:
Le cheratocisti odontogene hanno anche un tasso molto alto di recidiva. Questa è una delle loro caratteristica clinica principale. Questo tasso è ancora più alta nei pazienti con sindrome del nevo basocellulare e dista circa 82% contro il 25-60% nei pazienti sani (33). Ci sono tre meccanismi che spiegano le ragioni di questo alto tasso di recidiva:
- Escissione incompleta delle periferie della cisti oa causa di una parete molto sottile e friabile cisti, sia a causa di una perforazione della corticale ossea con l'adesione cisti di tessuto molle adiacente;
- Il verificarsi di cheratocisti odontogene da una cisti satellitare, un microcyst resti odontogeni dimenticati o dopo l'intervento chirurgico;
- Lo sviluppo di un nuovo odontogena cheratocisti in un'area adiacente e che è interpretato come una ricorrenza mentre è una formazione de novo che non ha legami con la lesione originaria. Secondo altri autori, il tasso di recidiva è anche influenzata da:
- Le dimensioni, posizione (Ramus), la morfologia della lesione (soprattutto multiloculaire) che rendono il trattamento più difficile e chirurgia
- La scelta della tecnica chirurgica (conservatori o aggressivi) La maggior parte delle recidive si trovano nei 5-7 anni del primo supporto.
Tuttavia, i casi sono stati riportati più di 10 anni dopo il trattamento. 25% dei casi ricorrenti si trovano da 9 anni in su. 54 Un aspetto essenziale del trattamento cheratocisti odontogene è monitorata nel lungo termine (dieci anni appare ideale per essere in grado di diagnosticare quasi tutti recidive in fase iniziale). Alcune caratteristiche di cheratocisti odontogene hanno portato l'OMS a riclassificare come tumori o neoplasie benigne piuttosto che le cisti: - primo comportamento: queste cisti sono localmente molto distruttivo, invasivo e hanno un alto tasso di recidiva; - Poi loro istopatologia: studi hanno dimostrato che le gemme strato basale nel tessuto connettivo. OMS ha inoltre osservato che mitosi figure si trovano spesso nello strato sovra-basale; epitelio cheratocisti un indice mitotico aumentata rispetto agli altri cisti mascellari (ma simile a ameloblastoma); - Per finire genetica: il gene PTCH è un gene soppressore del tumore localizzato sul cromosoma 9q22.3-q31. Questo gene è responsabile sia la sindrome del nevo basocellulare, ma anche responsabile della formazione di isolati cheratocisti odontogene. Codici normalmente PTCH per una proteina transmembrana che forma un ricevitore di segnale con l'oncogene SMO (lisciato) per ligando SHH (sonic hedgehog). Recettore SHH attiva questo quando c'è un'inibizione del segnale crescita di diversi tessuti e proliferazione. E 'coinvolto anche nella regolazione del metabolismo lipidico. Con contro, se si perde la PTCH operativo, avremo un effetto proliferazione cellulare (e quindi lo sviluppo di cisti e tumori).
Standard aspetto radiologico:
Molto spesso asintomatiche, la maggior parte delle cheratocisti vengono scoperti nel corso di un esame radiologico standard. Questo esame in nessun caso fare una diagnosi definitiva. Esso permette un orientamento della diagnosi può essere confermata solo dall'analisi istopatologica. L'aspetto a raggi X più comune di cheratocisti odontogena è il mono (di solito) o polygéodes radiotrasparenti, rotondi o ovali e con divisori interni sottili.
Nella mascella, sovrapposizioni con seno o cavità nasali può rendere l'identificazione di queste caratteristiche molto più difficile. Quando cheratocisti è molto ampia, il diradamento e la deformazione dell'osso corticale è possibile. Può svilupparsi su qualsiasi emi-mascellare; i condili sono sempre rispettati.
Quest'ultimo può anche avvolgere un dente non evoluta adiacente (incluso), un dente soprannumerario o odontome e causare lo spostamento o, molto raramente, il riassorbimento radicolare di altri denti si trovano a contatto.Periferiche elementi nobili (nervi ...) non sono distrutti, ma rinviate dalla cisti. La principale diagnosi radiologica differenziale si pone con ameloblastoma, cisti dentigerous poi alcuni.
4. Evoluzione
I casi di trasformazione maligna di cheratocisti odontogene in carcinomi a cellule squamose sono molto rari. Solo 12 casi sono stati riportati in letteratura fino ad oggi. Questa possibilità dovrebbe essere considerato quando ci sono più recidive, un comportamento molto aggressivo della lesione così come un fallimento di entrambi i trattamenti conservativi e trattamenti aggressivi. Si è spesso osservato nei pazienti anziani. Caso trasformazioni ameloblastico sono segnalati, ma anche molto rare
. 5. Diagnosi Differenziale
La diagnosi differenziale di cheratocisti odontogena deve essere fatto con altri tre cisti principalmente:
- con ameloblastoma:
Questa cisti è più frequente nei maschi e ha il suo picco di incidenza nel quarto e nel quinto decennio. 80% di ameloblastomas si trovano nella mandibola e soprattutto a livello angolazione e il ramo. Localizzazioni mascellari sono rari.
Da un punto di vista clinico, ameloblastoma è caratterizzata da una crescita illimitata e un alto tasso di recidiva.Queste lesioni hanno spesso un volume sufficiente. Generalmente asintomatici, sarà scoperto solo per caso di un esame radiografico. Altrimenti, può essere rivelato da un episodio infettivo o da movimenti e anomalie dentali evoluzione.
Da un punto di vista radiologico, ameloblastoma ha soprattutto un aspetto polygéodique "bolla di sapone", bordi regolari e con le partizioni interne. Geodi sono ben delimitate, rotonda o ovale, di varie dimensioni. Gli elementi nobili sono repressi, non distrutto. Ci sono tante somiglianze con il cheratocisti, rendendo la diagnosi differenziale impossibile con i soli risultati clinici e radiografici standard. La diagnosi differenziale sarà quindi principalmente mediante esame istologico, ma anche dalla TAC.
- con una cisti dentigera:
Questa cisti incontrato più frequentemente negli uomini ha il suo picco di incidenza nella seconda e terza decade.Tali disposizioni riguardano il terzo molare inferiore e il canino superiore e il secondo premolare inferiore.
Principalmente privo di sintomi, la cisti dentigera aderisce al collo di un dente e circonda la corona. La sua scoperta è più spesso nel corso di un esame a raggi X di routine o può essere motivata da movimento dentale inspiegabile.
L'aspetto radiografico è quello di un ben demarcata-osteolisi, monogéodique e sviluppato attorno alla corona o la corona di dente incluso e totalmente costruita. La lesione può diventare molto grande e invadere il ramo ascendente della mandibola. E 'più difficile da percepire radiologicamente inserendolo nel collo del dente incluso e quindi la differenziazione un cheratocisti.
Inoltre, è possibile osservare un riassorbimento della radice dei denti adiacenti alla lesione; che è molto più raro per cheratocisti odontogene ed è il principale segno distintivo radiologico di queste due entità.
- Con una cisti follicolari:
Questa cisti è una varietà di cisti dentigera ma che si sviluppa a contatto con follicoli dentali; vale a dire, i denti non completamente formate. La sua scoperta può essere effettuato sia mediante radiografia motivato da un ritardo eruzione di un dente permanente; sia durante un episodio infettivo (solitamente nella premolari). Altre caratteristiche sono identiche a quelle di Dentigerous cisti.
- Con una cisti infiammatoria banale o radiculo dentale:
Queste lesioni sono più frequenti mascellare. Essi preferenzialmente verificano in maschi nel mascellare anteriore o il ramo orizzontale della mandibola. Il loro picco di incidenza è tra il terzo e il quarto decenni. Allo stesso modo le lesioni visto in precedenza, la cisti radicolare è la scoperta di routine di solito asintomatica e radiologico. Può tuttavia essere rivelata in episodi infettivi caratterizzati da rigonfiamenti, gonfiore, pus accanto a un dente decaduto, necrotico o non completamente trasformati. Queste cisti sono a crescita lenta e possono, in assenza di segni clinici, espandere notevolmente.
Il loro aspetto radiografico è quello di un monogéode, arrotondata o ovale, in collegamento con l'apice del dente mortificato e circondato da una condensazione ossea confine (che possono scomparire durante attacchi infiammatori). Essi possono, in caso di rilevanti dimensioni, causa principale riassorbimento delle radici adiacenti. 59
La diagnosi differenziale può anche atterrare con altri cisti come il fibroma odontogeno, il mixoma odontogena, la cisti solitaria, angioma osseo o la cisti aneurismatica. 60 B.
B / Altri eventi odontostomatologiques
Altri eventi odonto- dentali sono anche incontrate nella sindrome del nevo basocellulare, ma con una maggiore disparità. Infatti, essi non sono sempre presenti, e quando sono, è con una frequenza molto inferiore alle cheratocisti odontogene che sono, essi stessi, uno dei principali criteri della malattia. Essi sono spesso direttamente legata allo sviluppo di cheratocisti e possono rappresentare uno dei primi segnali di chiamata per evidenziare loro (e quindi portare alla diagnosi di sindrome del nevo basocellulare)
un / Inclusioni o malposizionamento dentale
Essi riguardano principalmente la dentizione permanente. È possibile osservare una persistenza di denti primari quando il dente finale è trattenuta dal cisti, che impedisce la sua eruzione. Studi hanno dimostrato che, a seconda del tempo di sviluppo della cisti, le conseguenze livello dentale sono differenti. Infatti, quando il cheratocisti sviluppa quando costruzione radice, deformazione è stata osservata nelle radici come curvature o piega baionetta.
Se si sviluppa presto in contrapposizione ad una formazione elemento dentale, può provocare la lacerazione delle radici. Con contro, se la cisti si forma dopo l'eruzione dei denti, può essere responsabile per i viaggi e malposizionamento dentale, ma in ogni caso non può essere responsabile di riassorbimento radicolare o la polpa mortificazione.
b / prognatismo mandibolare
Si stima che circa il 25% dei casi. Può essere un vero prognatismo o pseudoprognathisme collegato a un brachymaxillie, o una combinazione di entrambi. 61 Il loro appoggio sarà attraverso la chirurgia ortognatica e ortodonzia. Prognatismo mandibolare in un giovane paziente con sindrome di Gorlin (15)
c / labbro e palato schisi
Il labbro leporino e / o palatoschisi sono presenti in circa il 5-7% dei casi di sindrome del nevo basocellulare. Una revisione della letteratura condotta da Lambrecht mostra che su 719 persone affette da sindrome di Gorlin, oro-facciale hanno 61 slot 8,5% dei pazienti. Queste persone possono essere suddivisi in due gruppi a seconda che lo slot è labiali, alveolare e palato (in 49 casi) o solo fessura (per 13 casi)
La frequenza di queste malformazioni non è la stessa tra la popolazione generale e nei pazienti con sindrome del nevo basocellulare
.Infatti, nella popolazione generale, l'incidenza di fessure labiali, alveolare e palato è che solo 1/800 e 1/2500 contro palatoschisi rispettivamente 1 / 14,7 e 1 / 55,3 di pazienti sindrome di Gorlin. Questa anomalia può essere uno dei primi indizi della presenza di una sindrome Gorlin in un neonato.
d ./ parestesia
Essi sono anche legati allo sviluppo di cheratocisti e si verificano quando la cisti raggiunge una dimensione significativa se si comprime o reprime un elemento nervoso e provoca parestesia. Può anche essere una complicazione postoperatoria di rimozione di una grande cisti con confini vicino nervo.
e / La améloblastome
Le ameloblastomas sono raramente riscontrati nei pazienti sindrome di Gorlin da pochi casi sono stati riportati in letteratura. Eslami mostra il caso di un paziente di 68 anni raggiunto dalla sindrome Gorlin ed avendo un ameloblastoma.
Mentre per quasi tutti i pazienti affetti da questa malattia, i primi sintomi compaiono in tenera età, di solito prima di 35 anni, i primi segni clinici del paziente cominciarono ad apparire molto più tardi.
In effetti, ha presentato la sua prima carcinoma basocellulare, all'età di 50 anni e più cheratocisti da soli 68 anni.Elemento interessante, abbiamo Eslami notato che 3 dei 4 casi di sindrome del nevo basocellulare associato con ameloblastoma registrato in letteratura inglese, anche presentato i loro primi sintomi in età più avanzata rispetto alla maggior parte dei casi di sindrome del nevo basocellulare.
L'età media per questi casi è 47,6 anni. Questa scoperta indica che i pazienti con sindrome del nevo basocellulare in età più avanzata sono più a rischio di ameloblastomas in via di sviluppo.
Come visto in precedenza, cheratocisti odontogene e ameloblastomas sono, entrambi, neoplasie benigne con un alto tasso di recidiva e un grande potenziale di distruzione ossea.
Tuttavia, ameloblastoma è più persistente e più infiltrante, che richiede una resezione ancora più radicale chirurgica.
sources :
