DR Gorlin RJ, SURGEON-HEKİM VE genetikçi, ONUN DERS
Bazal hücreli nevus sendromu GENETİK ÖZELLİKLERİ
A / GENEL
Bazal Hücre Nevus Sendromu KATILAN B / GENE
1. Genel
Çift olay 2. Teorisi
3. transdüksiyon Way yamalı / sonic kirpi
Aktörler iletim yolu / Sonic the Hedgehog yamalı
yamalı işletim / Sonic Kirpi iletim yolu
Yamalı yolu Sinyal / Kirpi Sonic
Mutasyonlar ve konumu Türleri
AYRINTILI OLARAK GENEL OLAYLAR Bazal hücreli nevus sendromu
1 / OLAYLAR Dermatolojik
A / nevus ve bazal hücreli karsinom Genel
B / palmoplantar hiperkeratoz Genel
C / Dermatolojik olaylar
2 / İSKELET ETKİNLİKLER
A / beyin kalsifikasyonlar zarflar
B / Anormal kafatası şekil
C / kaburga anormallikleri
D / vertebra anomalileri
El ve ayak E / Anormal kemik
F / Diğer iskelet bulguları
3 / GÖZ ETKİNLİKLER
Dünyanın A / Konjenital malformasyonlar
B / Tümör dünya süreci
4 / ETKİNLİKLER NÖROLOJİK
A / Méduloblastomes İstatistikleri
B / Diğer nörolojik belirtiler
5 / DİĞER GENEL ETKİNLİKLER
A / over myom
B / Kalp Myomlar
Erkeklerde C / hipogonadizm:
6 / ETKİNLİKLER ODONTOSTOMATOLOGIQUES
A / odontojenik keratokistler
B / Diğer olaylar odontostomatologiques
Bir / Kapanımlar veya diş malpozisyon
b / Mandibular prognatizm
c / dudak ve damak yarıkları
d / parestezi
E / améloblastome
BU NE Gorlin sendromu NEDİR?TANIMI
Ayrıca bazal hücreli nevus sendromu (NBC) olarak bilinen Gorlin sendromu, gelişimsel anormallikler bir dizi ve çeşitli kanserler için bir yatkınlık ile karakterize kalıtsal bir hastalıktır.
prevalans oran erkek / kadın 1, 000 000 1/57 ila 1/256 tahmin: 1.Klinik bulgular çok sayıda bazal hücreli karsinom (BHK), çenelerin odontojenik keratokistler, avuç içi ve ayak tabanı, iskelet anomalileri hiperkeratoz, intrakranial kalsifikasyonlar ve ektopik makrosefalisi yüzde dismor- (yarık dudak-palato varlığı, ve şiddetli göz anomalileri).Fikri açığı hastaların% 5'inde gözlenir.(1 ila 10 mm arasında değişen bir çapa sahip ülseratif plakalara cilt rengine sahip olan kabarcıklar itibaren) Bazal hücre karsinomları, genellikle, sırt ve göğüs yüzünde yer almaktadır.Bazal hücreli karsinom sayısı birkaç birkaç bin arasında değişmektedir.(Kaburga, omurga ve kafatası şeklini etkileyen) iskelet anomalileri sıktır.Ayrıca göz, genitoüriner ve kardiyovasküler bozukluklar meydana gelebilir. Gorlin sendromu olan hastalar arasında% 5-10 erken ölüm muhtemel nedenidir Medulloblastom, geliştirmek.
sendromu gen PTCH1 mutasyonlar neden olur ve tam penetrans ve değişken ekspressivite baskın otozomal olarak iletilir.Klinik tanı belirli ölçütlere dayanmaktadır.Tanı Araştırma mutasyonlar ile teyit edilir.Genetik danışma zorunludur.Antenatal tanı amniyosentez veya koryon villus örneklemesi ile elde edilen fetal hücrelerin ultrasonografi ve DNA analizi ile mümkündür.Ana ayırıcı tanı Bazex sendromu, multipl trichoépithélium ve Muir-Torre sendromu sendromu (bu terimleri bakınız) içerir.destek multidisipliner bir yaklaşım gerektirmektedir.
Keratokistler cerrahi rezeksiyon ile uzaklaştırılır.Lezyon sayısı sınırlı iken, bazal hücreli karsinom durumunda, cerrahi gerektirir.Diğer olası tedaviler lazer ablasyon, fotodinamik tedavi ve yerel kemoterapi vardır.
Radyasyon tedavisi kaçınılmalıdır.A vitamini analogları, yeni CBC gelişimine karşı koruyucu bir rol oynayabilir.Yaşam beklentisi önemli ölçüde değişmiş değil ama komplikasyonlar önemli morbidite neden olabilir.Multidisipliner bir ekip (dermatologlar, nörolog ve diş hekiminin) tarafından düzenli olarak izlenmesi gereklidir.Hastalar UV ışınlarına aşırı maruz kalmaktan kaçınmalıdırlar.
DR Gorlin RJ, CHRIRURGIEN- HEKİM VE genetikçi, ONUN DERS
Robert James Gorlin Hudson, New York 11 Ocak 1923 doğumlu ve 83 yaşında Minneapolis, Minnesota, 29 Ağustos lenfoma 2006 öldü.İkinci Dünya Savaşı sırasında orduya katıldı, o 1947 yılında mezun oldu o zaman Washington Üniversitesi'nde diş hekimliği eğitimi O Üniversitesi'nde Kimya Master alarak eğitimine devam etti Iowa State.Çok sayıda akademik pozisyonlarda sonra, 1956 yılında ölümüne kadar kaldı Minnesota Üniversitesi'nde profesör ve Oral Patoloji Bölümü Direktörü olarak bir pozisyon elde etti.
Gorlin achantosis nigrikanslı ile ilgili Helen Curth dermatolog bir kitap keşfeder kariyeri 1940 yılında bir hal aldı.Bu deneyim "syndromologiste" in Gorlin dönüştürmeye yardımcı olacaktır.achantosis nigricans cilt kalınlaşmış ve kareli, kaba görünen koltukaltı, boyun ve genital-femoral bölgede ağırlıklı olarak lokalize papiller hipertrofi ve pigmentasyon vejetan ile karakterize bir deri hastalığıdır.Bu dermatolojik durum gastrik adenokarsinom ile bir dernek ile karakterize malign formları vardır.Bu diğer vücut sistemlerini etkileyen bir dermatolojik hastalıktır.Bu keşif bazı sistemik hastalıklar ağız bulgularını sunmak eğer o istedi Gorlin büyüledi.Gorlin uluslararası birçok sendromların sözlü belirtileri üzerine yaptığı çalışmalarla tanındı.Kariyeri boyunca, Robert J. Gorlin ve arkadaşları okudu ve adlandırılmış yaklaşık 100 sendrom ve izolasyon ve bunların yaklaşık yarısından sorumlu genlerin karakterizasyonu katıldı.
Gorlin adı ortak bir ad, genetik hem de tıp değil sadece tanıdık 7 olmuştur.Bu onlardan biri olarak Gorlin dikkate dermatologlar, genetikçiler ve patologları istenir.Robert J. Gorlin çok üretken bir yazar olacak.Yaklaşık 600 yazılar, 60 kitap bölümü yazmıştır ve 20 kitap yazarlarından veya düzenlenmiş.Hatta bazıları, İspanyolca, Rusça ve Japonca çevrilmiştir.Gorlin adı da yaygın Gorlin sendromu olarak bilinen bazal hücreli nevus sendromu, gelen özellikle ayrılamaz hale gelmiştir.
1960 yılında, ilk kez kaburga deformiteler, kemik lezyonları ve kusurları ve göz tümörleri ile güneşte maruz kalmamış alanlar da dahil olmak üzere tüm vücutta görünen çok sayıda deri bazal hücreli karsinom arasındaki ilişkiyi açıklar .O dönem Gorlin sendromu altında bugün daha tek bir sendrom parçaları olarak bu anomaliler düşünmektedir.Bu anlayış ve nedensel genin, ptch genin bulunmasından yardımcı olacaktır.
Minnesota Üniversitesi'nde Kariyeri boyunca, geniş bilgi patoloji, dermatoloji, pediatri, kadın doğum, jinekoloji ve Boğaz otorhino dersleri vermeye yöneltti.O da Diş Araştırmaları Uluslararası Derneği, Oral Patoloji Amerikan Akademisi ve Cranio-yüz Biyoloji Uluslararası Derneği Başkanı oldu.Bilim, Tıp ve Toplum yaptığı büyük katkı için, Robert J. Gorlin Kariyeri boyunca birçok prestijli ödül aldı.Robert J. Gorlin nedenle onun çalışmalarıyla Diş Hekimliği yanı sıra tıp alanlarını ve genetik geçmeyi başardı bir eğitim diş hekimi vardır.
Bazal hücreli nevus sendromu GENETİK ÖZELLİKLERİ
A. GENEL
Uzun Gorlin RJ, terim bazal hücreli nevus sendromu olarak bilinen bu klinik tablo, daha önce zaten başka isimler altında, söz konusu açıklamaları olmuştur.Jarisch 1894 yılında
Kısaca skolyoz olan bir hastada iskelet anormallikler bazal hücre karsinomu kombinasyonunu tarif eder.Diğer hiçbir söz yapar
Hastalığın karakter.
Aynı yıl, Beyaz hayatı boyunca görünmeye devam multipl bazal hücreli karsinom, bir hastanın aile öyküsü bildirdi.
1939 yılında Straith bir ailenin üç üyesi birden fazla cilt bazal hücreli karsinom için maksiller kistler ilişkilendirmek için ilk.
1951 İki O dermatolog, Binkley ve Johnson, hem cilt karsinom ve maksiller kistleri tanımlamak kadar bekleyecek ve
Beyin değişir.
Bu Gorlin, diş hekimi ve Goltz, dermatolog bir sendrom düşünün bu klinik varlığın ilk kapsamlı tanımını yapmak gibi Minneapolis 1960 yılında oldu.Ona maksilla ve bifid kaburga çok sayıda kist ile bazal hücreli nevüs sendromu adını verecektir.
O zamandan beri, sendrom gibi Gorlin sendromu veya Gorlin-Goltz Gorlin-Goltz fakomatozdur, bazal hücreli nevus sendromu gibi çeşitli isimlerle tayin edilebilir ...
Bazal hücreli nevüs sendromu bilinen en eski vakalar nedeniyle dentigeröz kistleri, çatallanma kaburga ve Gorlin sendromunun diğer stigmata 1960'larda ortaya çıkarılmıştır Mısır toplama iki yetişkin erkek iskeleti iskelet üzerinde bulundu Torino Üniversitesi Antropoloji Enstitüsü.Bu iskeletler Hanedanlık dönemine Aşağı Mısır ve tarihi Assyut içinde mezardan bulundu.Hanedanlık dönemi kez iki krallık, Kuzey ve Güney bölünmüş Mısır birleşmesiyle başlar ve M.Ö. 3000 civarındadır.
bazal hücreli nevus sendromu otozomal dominant bir kalıtsal genetik bir hastalıktır.Ancak, sporadik vakalar bildirilmiştir: Olguların% 60'ında, başka hiçbir aile üyesi ulaşıldığında ve bu olguların% 35-50 néomutations temsil etmektedir.Bu nedenle aile öyküsü olmaması bu hastalığın tanısını ekarte etmez.Bu, bir gen olan 9q22.3-S31 bulunan ptch gen mutasyonu neden olduğu
tümör baskılayıcı.
nevoid bazal hücreli bir gen etkisini ortaya sıklığıdır yüksek penetrans sahiptir.Bu% 97 olduğu tahmin edilmektedir.Bu, bu% 97 anlamına gelir
Bu geni taşıyan bireylerin hastalık belirtilerinin en az birinden muzdarip.
Hastalık aynı zamanda değişken ekspresivitesini vardır;anlamlılık nicel değişken koşullarla şimdiki genin kalitesi olmak.Hastalık başka bir hastanın değişken çok belirtileri ortaya çıkmaktadır.Üzerinde kırk tanı kriterleri vardır.Bununla birlikte, bu hastalık, her bir hasta, tüm ölçütleri sunmazlar.Tanı kriterleri bağlı majör ve minör iki kategoriye içine sınıflandırılmıştır
oluşum sıklığı.Bu bazal hücreli nevus sendromu hastaları mevcut en az iki ana kriter veya ortakları tanı koymak mümkündür
İki küçük kriterlerde önemli bir kriter.
prevalans 3/1 dişi cinsiyet oranı erkek ile 1/56000 / olduğunu.Ghailan göre prevalans 1/60000 ve erkekler ve kadınlar etkilenir
Aynı oranlar.Bu cinsiyetler arasındaki eşitlik otozomal dominant sendrom ile çok iyi açıklanmıştır elde etti.
Wolff göre, prevalans 1/60000 den 1/120000 arasındadır ve her iki cinsiyette eşit etkilenir.Ayrıca sendromu gruplarının çeşitli oluşur
Kültür ve cilt tipine özgü bir yeğleme var görünmüyor.
Bazal hücreli nevus sendromu olarak şu anda sadece 700 vaka literatürde bildirilmiştir nadir bir hastalık olmaya devam etmektedir.Bununla birlikte, dişçi çene diş bulgular, bu hastalığın ilk belirtileri arasındadır, çünkü tanı koymak için birinci konumda olduğunda.
Bazal Hücre Nevus Sendromu KATILAN B. GEN
1. Genel
bazal hücreli nevus sendromu, ptch geni sorumlu gen, iki araştırma ekipleri tarafından 1996 yılında ilk defa tespit edilmiştir.Onun keşif oldukça yenidir
hastalığa yol açan tüm genetik mekanizmalar iyi bilimsel topluluk tarafından anlaşılmış değildir, bazı unsurlar belirsizliğini koruyor.
Kromozom 9'un uzun kolu üzerinde yer alan ve daha hassas 9q22.3 olarak, bu gen Gorlin sendromu olan hastalarda değil, aynı zamanda durumlarda mutasyona uğramış halde bulunduğuna
sporadik bazal hücreli karsinom.
Bu gen, embriyo gelişiminde önemli bir rol ve hücre çoğalmasının kontrol oynar.
1982 bir makalesinde, Fitzpatrick bu sendromun kökenini açıklamak için geçerli verileri çok farklı bir teori sağlar.Zaman bilim adamları teknikleri, aslında, bugün bildiğimiz genetik anomali tespit için başarısız oldu.O çok sistemik anomaliler enzimatik defekt varlığını açıklamak söyledi.Gebeliğin ilk üç aylık döneminde böyle bir kusurun varlığı iskelet anomalileri ve kalsifikasyonlar zarf beyin açıklayabilir.
Daha sonra, deriyi kontrol enzimatik mekanizmasında bir anormallik nevüs, bazal hücreli karsinom, palmar-plantar çukurlar ve keratokist oluşumuna yol açabilir.Böyle medulloblastoma gibi nörolojik tümörlerin kökenini anlamak için permetpas yapar Ancak bu teori yetersizdir.
Çift olay 2. Teorisi
Bazal hücreli nevus sendromu çalışma tümörleri ptch geni bir tümör baskılayıcı geni olabileceğini düşündüren, 9q bölge için heterozigotluk kaybı varlığını gösterdi.Olan prototip zaman retinoblastoma geni (Rb) anti-onkogen Bu düşünce, Knudson tarafından önerilen modelden bir tümörde bu genin her iki alel kaybı (heterozigozite kaybı 13) dayanır İki mutasyon olayları 'onun "teorisine göre 1971 yılında, ilk mutasyon malformasyon sendromu sorumludur ve tümörlerin oluşumuna zemin hazırlayabilir ve ikinci tümörlerin ortaya çıkmasına yol açacaktır.
Bir tümör baskılayıcı geni inaktive olmakta, böylece Cohen göre, iki fenomen gerekmektedir.İlk bir allelin aktarılmasıdır.İkinci olgu heterozigosite (LOH ya da) kaybı olarak bilinen diğer alel, kaybıdır.Her iki allel kayıp olduğunda, tümörler oluşur.Heterozigotluk kaybı temel özellikleri Gorlin sendromunun üç olan bazal hücreli karsinom, odontojenik keratokistler ve medulloblastom için gösterilmiştir.
Böylece, ailesel formları Gorlin sendromunda, ilk mutasyon anlayışı mevcut olabilir ve tüm vücut hücrelerinde bulunur.Yavruda bazal hücreli nevus sendromu geliştirmek için bir yatkınlık yaratan bu ilk mutasyon olay, baskınlık iletilir.Bu olay gizli kalır ve sadece ikinci olay kromozomu üzerinde oluşursa anlaşılmaktadır
görünüşü Gorlin sendromu ile sonuçlanan aynı mevkide homolog.(Genetik malzemenin ilk kalıtsal beri) ailesel durumlarda, ayrı ayrı tek bir mutasyon adımı gerekmektedir.Hiçbir aile öyküsü olan bir bireyde görünüm Gorlin sendromu karşı tarafından
Aynı bireyde iki ardışık mutasyon olayları meydana gelmesini gerektirir.
3. transdüksiyon Way yamalı / sonic kirpi
Ptch geni Drosophila yamalı genin insan homologudur.Bu gen yolunda meydana gelen Drosophila geni (veya polarite) bir segmentasyon olduğunu
embriyonik gelişim ve hücre çoğalmasının kontrol katılır transdüksiyon yamalı / sonik kirpi.Bu ligandın yokluğunda hücre bölünmesini durdurarak ve bağlantı ligant ile yapılır kısa aktive ederek hücre döngüsü düzenleyicisi olarak işlev görür.
Gorlin sendromu beyinde hasara neden olur, kaburga, omurga, üyeler ... Yamalı bu dokuların boyunca ifade edilir.
Aktörler iletim yolu / sonic kirpi yamalı:
Büyük ilerleme sinyal yolağı yamalı / sonik kirpi çalışmasını anlamakta son yıllarda yapılmış olmasına rağmen, birçok
belirsizlikler hâlâ gelecekte açıklığa kavuşturulması gerekmektedir.
- Ptch geni tarafından kodlanan Patched phenylthiocarbamide ya da protein, on iki transmembran alanları ile 1447 amino asitten oluşan bir glikoproteindir,
iki büyük Hücre dışı düğümler da ligand SHH, geniş bir hücre içi döngü, bir hücre-içi amino-terminal etki alanı ve bir karboksi terminal hücre içi bağlanması için gerekli.
Bu bazal hücreli nevus sendromu mutasyona uğramış ama aynı zamanda bazal hücreli karsinom ve medulloblastom nadir durumlarda görülür.
En çok mutasyon, bir kesik proteini sentezine neden olmayan mutasyonların bulunmaktadır.(Lacombe) Gene 1 yamalı
- Sonic dikenli proteini (Shh), 7q36 Shh bulunan gen tarafından kodlanan.Bu 45kDa proteini, iki etki alanı içine bölünür: 20 kDa, Zn, bir amino-terminal alanı, bir hidrolaz aktivitesi ile donatılmış 25 kDa otokatalitik bir karboksi-terminal sahası olan
Kolesterol transferaz aktivitesi.
Bu sinyal yolunun kolesterol rolü tam olarak bilinmemektedir ancak SHH molekülünün ve dağıtım yayılmasını sınırlayan bir rol oynayabilir
mekansal etkisi.
- Son olarak, 7q31-32 düzeltilir bulunan gen tarafından kodlanan Smoothened protein (SMO).
Bu (etti Wnt reseptörlerine homoloji göstermektedir), G proteinlerine bağlanmış serpantin alıcıların familyasına ait 787 amino asitten oluşan 115 kDa'lık bir proteindir.ikinci sinyal katılan bir hücre dışı amino-terminal etki alanı, bir karboksi-terminali hücre-içi etki alanı ve iki alanları (üçüncü hücre içi halka ve yedinci transmembran alanı) sahiptir.
yamalı işletim / Sonic Kirpi iletim yolu:
SHH rolü ligandı için bir alıcı olan yamalı.Ve SHH'nin, SMO Patched yokluğunda inaktif kompleks oluşturur.Bir şekilde Patched eylemler
negatif regülatörüdür, SMO bir inhibitörü.
SHH Patched bağlanan söz konusu olduğunda, ikinci daha sonra kapatılır ve SMO hücre içinde bir sinyal iletmek için serbesttir.
Birkaç mekanizma SMO ve sinyal iletimi aktivasyonunu açıklamak için önerilmiştir:
- Birinci hipotez olarak, Yamalı ve SMO bir kompleks oluşturur.SHH Patched / SMO kompleksinin ikinci ve dolayısıyla ayrışma konformasyonel bir değişim ile reseptörünün Patched sonuca ligand bağlanmasını.SMO sonra hücre içinde sinyal iletmek için ücretsiz olacaktır.
- Daha yakın zamanda, ikinci bir hipotez, düzeltme eki ile stoikiometrik olmayan düzenleme önermektedir.Gerçekten de, testler sonucu diminutionde Patched SHH'nin bağlanma gösterdi ve hücre zarında GOS artmıştır.ilişkisi
atched ve SMO 16 Yamalı düzenleyecek stokiyometrik olmayan, yani olurdu
SMO değişim ama onunla gerçek bir kompleks teşkil etmemektedir (ikisi arasında hiçbir fiziksel bir etkileşim var olacaktır).
SMO devreye girer ve çekirdeğe sinyal iletimini gerçekleştirebilirsiniz.
GOS ile hassas bir sinyal iletim yolu henüz bilinmemektedir, ancak bu son hedef transkripsiyon faktörü GLI (kubitus interruptus homologu veya Cı Drosophila) aktivasyonu ile temsil edildiği bilinmektedir.Hücrelerde SHH GLI için maruz kalmayan bu Kıyı-2 (cos2) Fused (Fu) ve Erime faktörü (Sufu) supresör gibi diğer proteinler ile bir kompleks oluşturur tétramétrique.Bu formda, GLI nükleuslar transloke edilebilir ve ikinci hedef genlerinin baskı yapabilen 75 kDa amino terminal parçasına dönüştürülmek üzere bölünebilir.
SHH ligandın varlığında, kompleks ayrıştığında ve bütün rolü GLI transkripsiyonel aktive oynar (çekirdek oluyor ve hedef genlerin ekspresyonunu sağlar).
Hedef genler:
- Kanatsız (WG) omurgalılarda WNT ailesinin kodlama üyeleri,
- Decapentaplegic (DPP) kodlayan, kemik morfojenik protein (BMP) ve TFG beta
- PTC kodlama kendini yamalı.
Bu genler, normal embriyonik gelişme ve bazı dokuların farklılaşması için çok önemlidir.
bu farklılaşma, göç, hücre çoğalması ve kutupsallık gelişim süreçleri dahil glikoproteinlerin bir ailesi için kanatsız geni kodlar.Bu genin ekspresyonu, düzensiz gelişim anormallikleri ve onkojenez sorumludur.
decapentaplegic gen embriyonik ektodermin dorsoventral farklılaşma bilgi iletimi küçük bir rol oynamaktadır.
Yamalı yolu Sinyal / Kirpi Sonic
Gelişiminde rol Patched:
Bir kirpi sinyal yolu dokularının çeşitli kök hücrelerin gelişiminde önemli bir rol oynar.Ptch hem de ifade edilir
o doğum sonrası dönemde rol devam ettiğini düşündürmektedir embriyonik gelişim ve erişkinlerde.Uygunsuz kirpi sinyal yolunun herhangi bir bileşenini ifade eden deneysel modeller önemli gelişimsel anomalileri ortaya koymaktadır.Bu anormallikler ve bu özellikleri, Gorlin sendromu arasındaki benzerlik bu sinyal yolunun aşırı aktivasyonu lacnaevomatose bazal hücrede bulunan gelişim anormallikleri ve tümörlerin neden olduğu anlamına gelmektedir.
SHH'nin tarafından teslim sinyalin iletim düzenler Yamalı gibi ventral nöral tüp oluşumu gibi birçok embryonaux süreçlerde dahil sinyal, beyin, bacaklarda veya diş gelişimi parçaları ...
Mutasyonlar ve Yer Türleri
Pek çok mutasyon, yazarlar tarafından rapor edilmiştir.Nitekim, bazal hücreli nevus sendromu, tek bir mutasyon isnat edilemez.Hastalara göre, ptch geninin tüm kodlama dizisi üzerinden dağıtılabilir olmayan mutasyonların, yanlış anlamlı, ters silme ... vardır.
mutasyonların çoğu, kesik bir proteinin sentezine yol açan bir durdurma kodonu yaratılmasına yol açar.
Bu mutasyonlar, geniş hücre içi döngü, geniş bir hücre dışı döngü ve ptch genin amino terminal bölgesinde çoğu için konsantre edilir.
Hiçbir ilişki genindeki mutasyon konumu ve farklı hasta Gorlin sendromunda gözlenen fenotip arasındaki kurulabilir.Bu sendromun bir özelliği genotip / fenotip ilişkisinin olmaması.Aynı mutasyonu taşıyan bireyler için, klinik resimler çok değişken olabilir.Bu durum, hastalığın gelişmesinde çevresel veya genetik faktörler gibi diğer faktörlere etkisi olasılığını göstermektedir.
AYRINTILI OLARAK GENEL OLAYLAR Bazal hücreli nevus sendromu
1 / OLAYLAR Dermatolojik
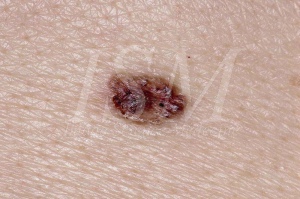
A / nevus ve bazal hücreli karsinom Genel:
Bazal hücreli nevüs bazal hücreli nevüs sendromunun ana alametlerinden biri olacak şekilde bu nevus yokluğunda;Bazı doktorlar teşhis amblemi kaldırın.Onlar keratokist odontojenik aynı şekilde, hastalık için en önemli kriterlerden biridir.Onlar bazal hücreli karsinom, malign dejenerasyon büyük bir evrimsel bir potansiyele sahiptir.
Onlar Gorlin sendromunda vakaların% 90 üzerinde bulunur.Bu bazal hücreli karsinom veya daha fazla 200 hasta Gorlin sendromu ulaşıldığında tahmin edilmiştir.Daha bazal hücreli karsinom tanısı yapılır er bu oran artar.20 yaşından önce bazal hücreli karsinom ile yaklaşık bir beş hastada bazal hücreli nevus sendromu muzdarip olduğunu düşünüyor.
Başlangıç yaşı:
Bazal hücreli karsinom, bölüm Gorlin sendromu gibi, normal popülasyona göre daha erken yaşta görülür.Onlar doğumda mevcut olabilir veya çocukluk döneminde gelişebilir.Çoğu zaman, onların oluşum ergenlik ve kırk yaşına başlar, bu hastalıktan etkilenen hastaların% 90'ı en az bir bazal hücreli karsinom geliştirdik.
Numara: bazal hücreli karsinom sayısı çok değişkendir.Klinik durumlar tek kanserli görülmemiş olmasına rağmen, çoğu durumda, sayısı yirmi ve binlerce arasındadır.
Yer:
Bu tümörlerin yeri maruz kalmayan alanlarda daha güneşe maruz kalan bölgelerde (ve dolayısıyla ultraviyole ışınlarının) hem yapılabilir.göz kapakları, burun, yanaklar ve alın en fazla etkilenen siteler ama boyun, gövde ve aksilla da sık sık etkilenir vardır.Eksileri, saçlı deri ve uzuvlar vardır tarafından, çoğu zaman bağışladı.Klinik görünüm: Bazal hücreli nevüs değişken klinik yönlerini alabilir.
Üç yönleri genel olarak tarif edilmektedir:
- Molluscum contagium çağrıştıran Görünüm bulaşıcı cilt durumu ve belirli koşullar altında aşılanmış olduğunu. Bir virüsün neden olduğu ve mat beyaz küçük yükselmeler ile karakterizedir veya inci boyutunu gül ve üst göbek bir depresyon büyümüştür.
- Hiperpigmente büyük nodülün Görünüm.
- Klasik bazal hücreli karsinom çağrıştıran Görünüm. Bu en yaygın olanıdır. Nevüs sonra küçük yumuşak tümör saydam yüzeyli olarak görünür. Onun renklenme, genellikle bu normal deride, bazen soluk pembe veya gri değişkendir. Tutarlılık görünüm beklenir çok daha güçlüdür, ancak baz sızmış değildir. boyutu çap olarak 1 ila 3 mm arasında değişir. Bu lezyonların herhangi bir kabuk ile kaplı boyutta artmış veya kanamaya başlarsa veya olursanız, malign transformasyon şüpheleniliyor. Bazal hücreli nevus sendromu içinde görünen Bazal hücreli karsinom açıkça bu sendrom hasta tarafından bir etkilenmeden görünmesini olanlardan ayırt edilemez. Sadece karakteristik unsur Gorlin sendromu çok sayıda ve erken yaşta ve güneşe maruz kalan bölgelerde zorunlu değildir konumlarına, kendi olaydır. Bu tümörlerin klinik görünümü kahverengi ten rengi papül veya ülseratif plaklar arasında değişen oldukça değişkendir. Onlar nevüs veya hemanjiom için yanlış olabilir. Bunların boyutu çap olarak 1 ila 10 mm arasında değişir.
Histoloji:
Bazal hücreli nevüs histolojik görünümü çevresel çit oturan çoğu kez, o iyi tanımlanmış aralıkları oluşturan epitel hücrelerinin bir intradermal çoğalması, biridir. Altta yatan stroma Fibroblastlarda zengindir. bazal hücreli karsinom hastaları Gorlin sendromunun histolojik görünümü olmayan sendromik hastalarda bulunanlara farklı değildir. Nevüsler bazal hücreli karsinom, bir uyarilar büyük ve net hücreleri mitoz artan sayıda, infiltratın inflamatuar karakterini olduğunda. Bu lifli stroma sahiptir. Lezyon papullaire ya saplı olabilir. Derin girmeler, ülser ya da işgaller lenfositik infiltrasyonu ile oluşabilir. Bunlar, kütle ya da periferde pigment olabilir.
Güneşin Etkisi:
Bir çalışma bazal hücreli nevus sendromu olan hastalarda ultraviyole ışınlarına karşı artan duyarlılık sunacaklarını göstermektedir. Bu kuzey-batı İngiltere'de Gorlin sendromu olan hastaların sadece% 14 Avustralya'da% 47 karşı, bazal hücreli karsinom gelişebilir gerçeği tarafından desteklenmektedir. Bu tanı sırasında UV maruziyetini sınırlamak için (olgu de novo çıktı için) (tek ebeveyn ulaşıldığında) prenatal tanının önemini ya da mümkün olan en erken tanıyı vurguluyor Pozitif.
Başka bir çalışmada, bazal hücreli karsinom ve kadınlarda genel popülasyonda erkeklerde% 86% 88 kadınlarda% 59 ve bazal hücreli nevüs sendromu erkek taşıyıcıları arasında% 65 karşı güneşe maruz kalan vücut bölgeleri üzerinde meydana raporları taşınan . O lezyonların anatomik dağılımı, güneş bazal hücreli karsinom gelişiminde önemli bir faktör olmadığını göstermektedir sonucuna varmıştır. Maruz kalan bölgelerde fazla sayıda Onların varlığı, ancak, güneş bazal hücreli karsinom gelişimini arttırabilir ve kendilerini korumak gerektiğini düşündürmektedir. O bazal hücreli karsinom sayısı ve güneş ışığına maruz kalma saatleri sayısı arasında anlamlı bir ilişki rapor vermedi.
Sadece Gorlin sendromu olan Afrikalı Amerikalıların yaklaşık% 40 Kafkasyalıların% 90 karşı bazal hücreli karsinom gelişebilir. Gorlin sendromlu 105 hasta çalışma Afrikalı Amerikalıların sadece% 38'i Kafkasyalıların% 97 karşı bir veya daha fazla bazal hücreli karsinom olduğunu göstermektedir. Dahası, Kafkasyalılar daha erken bazal hücreli karsinom gelişebilir. Nitekim, Kafkasyalıların yaklaşık% 50'si 21.5 yıl ilk karsinom gelişebilir ve Afrikalı-Amerikalılar aynı yaşta% 20 ve% 40 karşı 35 yıl 90%
Afrikalı-Amerikalıların grubunda en küçük frekans bazal hücreli karsinom nedeniyle cilt pigmentasyonu nedeniyle ultraviyole ışınlarına karşı daha iyi koruma olabilir.
İyonizan radyasyon etkisi:
Iyonize radyasyon Bu tümörlerin gelişiminde rol oynadığı gösterilmiştir. Nitekim, radyoterapi tedavisi gören hastalar genellikle ışınlanmış bölgelerde çok sayıda bazal hücreli karsinom vardır. Onlar 6 ay ve Gorlin sendromlu olmayan bir hasta ile 21 yıllık ortalama karşı ışınlama sonra 3 yıl arasında bir sürede ortalama salınım görünür.
Bu hastalığın erken belirtilerinden biri medülloblastom olduğu unutulmamalıdır. Şimdi genel olarak radyoterapi ile muamele edilmektedir. Işınlanmış alanda BCC gelişme riskinin, kuvvetli şekilde arttırılır. Buna ek olarak, bu karsinomları daha agresif ve bu nedenle daha zor tedavi bulunmaktadır. Bu nedenlerden dolayı, biz radyoterapi tedavileri en aza indirmek ve cerrahi tedavi teşvik etmelidir.
Evrim:
Bölüm Gorlin sendromu, daha agresif tedavi edilmesi daha zor gibi görünen olarak Bazal hücreli karsinom görünür. Onlar tedaviye veya direnç (beyin ve özellikle akciğerlerde metastaz) istilası hastanın ölümüne neden kadar çıkabiliyor. Malign transformasyon çoğunlukla ergenlik sonrası görülür. en agresif tümörler en sık göz kapaklarını ve burnu etkileyen ve hastaların önemli bir şekil bozukluğu neden olabilir doku hasarına neden olur.
B / palmoplantar hiperkeratoz Genel:
Onlar da "çukurlar" koşulları altında bir araya geldi, delik veya kuyu palmoplantar. 26 Frekans: Onlar vakaların yaklaşık% 65 ila% 80 mevcut olduğundan erken yaşta meydana gelen, onların varlığı bazal hücreli nevus sendromu erken tanısında önemli bir yardımcı olabilir. Onlar tüm yaş gruplarında hemen hemen aynı frekansta ile bir araya geldi. Palmar birkaç dakika su içine daldırıldıktan sonra el hiperkeratoz.
Klinik görünüm:
Bunlar kısmen ya da tamamen yok olduğu stratum corneum içinde çok biçimli kusurları küçük deliklere karşılık gelir. Kir koyu iç, birikmiş eğer Nadiren çapı 1 ya da 2 milimetre ve derinliği 1 mm üzerinde kendi arka plan, pembe ya da olabilir. Bu lezyonlar sadece ellerde ve ayaklarda karşılaşılan asemptomatik vardır. Klinik tanı zor olduğunda, uygulayıcı ılık suda birkaç dakika el ve / veya hastanın ayak sokmak olabilir. çukurlar sonra daha net görünür.
Histoloji:
Bir avuç çukurlar histolojik incelemesi, küçük bir ön proliferatif bazal hücreler gösterdi. Bu önünde yer alan bir hücresel düzensizlik olsa da, ön ve bitişik stroma arasındaki çatlak görülmemiştir. Hafif damar çoğalması ve fibrozis komşu dermis gözlendi. istologique
Sonuç olarak, palmoplantar çukurlar karakteristik belirtisi Gorlin sendromu bulunmaktadır. Onlar yüzünden oluşum ve gerçekleşme erken yaşta yüksek frekans bu sendromun tanısında önemli bir yardım vardır. Bu nedenlerden dolayı, onlar bazal hücreli karsinom ve keratokist ile hastalık için en önemli tanı kriterleri arasında yer almaktadır.
% 75, çalışmaya bağlı olarak 35 ve bazal hücreli nevus sendromu ile ilişkili olarak deri kist frekansıdır. Epidermoid kistler genellikle vücut pilleuses alanlarına ve nadiren böyle avuç içi ve ayak tabanlarında olmayan pilleuses alanlarda görülür. Vücudun ucunda bu tipik konumu bazal hücreli nevus sendromu farklı bir klinik bulgusu olabilir.
Bu histolojik açıklama keratokist, bazal hücre nevus sendromu önemli bir tanı kriterleri ile çok benzer. Bunlar deri keratokist histolojik çenelerin keratokistlerin yakınları, bazal hücreli nevus sendromu tipik bir marka olabilir.
, Gorlin sendromu tanısı doğru bize yol açabilir "klinik inci" olarak kabul edilmelidir Sonuç olarak, özellikle ellerde ve ayaklarda bulunan deri kistlerin bulunması, bazı keratokist ile güçlü bir benzerlik histolojik var .
c / Diğer dermatolojik bulgular
Diğer dermatolojik bulgular literatürdeki bulunur, ama nedeniyle oluşum çok düşük frekansa açıkça tarif edilmez. Hastalar böylece lipom, tahıl milia, leke "latte" ya da Molluscum bir sarkaç olabilir. Sağ skapula üzerinde bulunan Lipom. Ayrıca çok sayıda deri nevus varlığı dikkat. yumuşakçalar sarkaç fibröz tümörler ve sarkık deri vardır. Onlar hissediyorum depresyonda ya da tersine, projeksiyonlar ve hatta saplı, düzlemsel ve kaplama olabilir. milia küçük yüksekliklerde oluşumu ile karakterize bir cilt nadir bir hastalıktır parlak saydam, üst tabakaların dejenerasyon kolloid (jöle bir tür hücrelerin transformasyon) tarafından üretilen bir topluiğne başı büyüklüğünde Dermis.
2 / İSKELET ETKİNLİKLER
A / beyin kalsifikasyonlar zarflar
Bu fizyolojik çocuk veya patolojik intrakranial kalsifikasyon karşılamak mümkündür. Eski çok nadirdir. İkincisi daha sık görülen ve genellikle tümör (medulloblastom gibi), nöro-kutanöz sendromlar, enfeksiyon ve nörodejeneratif bozukluklar ile bağlantılı olarak tarif edilmiştir. Genel popülasyonda, intrakranial kalsifikasyonlar Gorlin sendromu olan hastalarda, yaşamın ilk yıllarından itibaren görünür, oysa yaklaşık 12 yıl ortalama görünür. Bu nedenle bazal hücreli nevüs sendromu tanısı için erken göstergesi olarak kullanılabilir. Genellikle iki hemisfer arasında merkezi bir bölüm oluşturan dura mater parçası olan sahte beyne, ilgilidir. Hastalar Gorlin sendromunda frekans 65 ve yazarlara göre% 85 arasında gidip beri oldukça önemlidir kanıtlıyor. Bunlar en sık radyolojik işaret etmektedir. Tentoriumun Kalsifikasyonlar da yanlış beyin üzerinde çok ama çok daha nadirdir. beyincik, beyin ve beyincik çadır arasındaki dura mater parçasıdır. Aynı zamanda korpus kallosum agenezisi ile bir araya mümkündür ancak bu anomali nadir kalır.
B / Anormal kafatası şekil
Bazal hücreli nevus sendromu olan hastalarda kafatası şekil anormallikleri var. Bu anomaliler hastalığın minör kritere ait olmasına rağmen, büyük ilgi tanı onlar doğuştan görünür çünkü. Bazı hastalarda kafatası frontal bossing ve parietal-zamansal büyük önem anormal olduğunu. kaşları belirgindir. hipertelorism (gözlerin aşırı boşluk) yaygındır ve prognatizm bazen bildirilmiştir. burun tabanı çöktü ve genişlemiştir. Bir burun septum sapması da Gorlin sendromlu 105 hasta çalışma izlenebilir, makrosefali, normal popülasyona göre bu hastalarda daha sık (olguların% 50) ile görülmektedir. Bu durumda,% 42 oranında mevcut olan hypertélorisme için aynıdır.
C / kaburga anormallikleri
Hastalığın minör kriterlere ait olmakla birlikte popülasyonda frekans çok düşük ise, kostal anormallikler hastaların yaklaşık% 42 mevcuttur. Bunlar büyük ölçüde mevcut ve çoğunlukla temsil edilir: - bifidités - agenezi - kıyı sinostoz. sinostoz iki komşu kemiklerin toplam kaynak olduğunu. Bu kaburgaların ön veya arka ucunun anormal genişlemesi gözlemlemek mümkündür. üçüncü, dördüncü ve beşinci kaburga en yaygın tüm kaburga elde edilebilir olsa bile, bu kusurlar etkilenir. Kimonis gibi bazı yazarlar, iki nedenden dolayı hastalığın başlıca kriterleri arasında kosta anomalileri sınıflandırmak karar verdi. İlk genel nüfusa oranla hastaların yüksek frekanslı (% 42) 'dir. İkinci Bu anomaliler kolayca doğumda tespit ve dolayısıyla pediatrik popülasyonda bazal hücreli nevus sendromu önemli bir tanı elemanı temsil edilebilmesidir. Kimonis kriterlerini uygulanması, hastalığın erken bir teşhis hastaların çok sayıda gerçekleştirilebilir. Nitekim, bu tür odontojenik keratokistler veya birden fazla bazal hücreli karsinom gibi hastalık için diğer önemli kriter, doğumda mevcut değildir; Bunlar çoğunlukla çocukluk ya da ergenlik döneminde görülür. Klinisyenler çocukluk döneminde hastalık minör klinik belirtileri çok dikkatli olmalıdır. Gorlin sendromunun varlığı kaburga anomalileri, kalp fibrom, damak veya yarık dudak, polidaktili veya makrosefalisi doğan çocuklarda aranmalıdır.
D / vertebra anomalileri
Gorlin sendromu olan 105 hasta çalışmaya göre, skolyoz vakalarının daha sık kişilik grup nasıl etkilediğini tespit edildi. Bununla birlikte, bu fark, anlamlı olması için yeterince büyük kanıtlamamaktadır. Hasta bireylerin daha şiddetli genel nüfusa göre daha skolyoz ve en sık bu tür hemi-vertebra füzyonları veya vertebralarda olarak gelişimsel anormallikler sitelerinde bulunan var görünüyor. Bu gelişimsel anomalileri olguların% 31'inde rastlanmaktadır. Spina bifida vakaları ancak genel nüfusa göre biraz farklı bir frekansta bildirilmiştir. Spina bifida az ya da çok büyük tümör şeklinde fıtık olan ile bir veya daha fazla omurun varsayılan omurga kemikleşme noktalarının, lehim arka ekseninin bir çatlak içinde oluşan bir kusurdur, zarları ve beyin omurilik sıvısının değişken miktarda bazen iliği. Servikal ve torakal sütunlar hastaların çoğunda Gorlin sendromunda bu kusur etkilenen
El ve ayak E / Anormal kemik
Polidaktili bu vakaların sadece% 3 karşılaşılan gibi, ancak kolayca saptanabilir ve doğumdan mevcut çok yaygın bir deformite değildir. Onun tanı kolaydır. Hastalar Gorlin sendromu da dördüncü ve / veya beşinci metakarp bir kısalma olabilir. Onlar seyrek çünkü bu malformasyonlar hastalığın minör kriterleri arasında bulunmaktadır.
F / Diğer iskelet bulguları
Sprengel deformitesi: bazal hücreli nevus sendromu olan kişilerin% 11 skapula doğuştan deformite Sprengel veya yükseklik var. Bu anomali yerinden kemik omurga yukarı ve içe deformasyon ve bazen de eki ile bir veya her iki skapula hareket içerir. Bu artış nedeniyle fetal hayatın boyunca kemik normal iniş pozisyonu bir başarısızlık etmektir.
3 / GÖZ ETKİNLİKLER
göz tutulumu sıklığı bilinmemektedir. Bu hastalığın en yayınlanmış çalışmaların önemli kriterleri tedavi onlar, açıklama Gorlin sendromunda az yer kaplar, çünkü budur. Bir söz konusu değil, gerçek bir açıklama değildir Göz bozuklukları.
Dünyanın A / Konjenital malformasyonlar
karşılaşılan başlıca göz kusurları katarakt, glokom ve kimin frekansları kombine Gorlin daha az% 14 olduğu tahmin edilmektedir coloboma vardır.
Katarakt lens donukluk ya da kapsülün bu yol açan bir göz bozukluğudur.
Glokom 20 mm Hg üzerinde artış göz basıncı ile karakterize bir göz durumdur. Bu sulu salgının normal akıştaki rahatsızlık kaynaklanmaktadır.
Kolobom tek veya birden fazla çevresel çentik oluşan lens kusurdur.
bazal hücreli nevüs sendromunun tipik yüz özelliklerinin bir parçası olarak gözler arasındaki hipertelorizmi veya aşırı mesafe, aynı zamanda sık sık karşılanmaktadır.
Bu anomalilerin yanı sıra, bu şaşılık, konjenital katarakt, mikroftalmi veya retinitis pigmentosa vakalarını bulmak da mümkündür. Retinitis pigmentosa retina, bilateral, aile ve kalıtsal dejeneratif bir süreçtir. Bu körlüğe sonunda lider, çocukluk çağında görülen ve görme keskinliği ve görme alanı daralmasına ilerleyici kaybı ile karakterizedir
B / Tümör dünya süreci
göz kapakları sıklıkla malign transformasyon riski taşıyan bazal hücreli nevus koltuk bulunmaktadır. Bu dönüşüm bir torpid ülserasyon kurulmasıyla tezahür edilebilir invazif veya kapak sünneti ile derinlemesine infiltrasyonu ve (o gelişmeye değildir ve gerileme değil, hangi söylemektir). Göz kapaklarında Bazal hücreli karsinom genellikle daha agresif ve tedavi etmek zordur. Dahası, onların tedavisi hasta için önemli bir yüz sünnetini neden olabilir.
Olgu de bildirilmiştir Salazyon. Salazyon göz kapaklarında çerçevesini oluşturan, tarsal kıkırdak bir meibomian bezi, yağ bezinin kronik inflamasyon küçük bir gözkapağı tümördür.
4 / ETKİNLİKLER NÖROLOJİK
A / Méduloblastomes İstatistikler:
Genel popülasyonda, medulloblastomlar çocukluk çağında en sık görülen beyin tümörleri ve tüm bu tümörlerin yaklaşık% 20 etkiler. ortalama tanı yaşı 6 yıl 3 ay ve erkekler kızlara göre daha büyük ölçüde etkilenir. Bu tümörlerin geleneksel tedavi radyoterapi ve kemoterapi ile kombine cerrahi rezeksiyon oluşur. 5 yıllık sağkalım oranı% 60 ila 80 arasındadır. Yaklaşık Medulloblastomlu 5 yıldan az Hastaların% 5'i Gorlin sendromu ile elde edilir. Bu oran, yetişkin durumda 1 veya% 2 oranında düşer. bu 2.1 yıl olduğundan hastalarda yaş ortalaması medulloblastoma tanı erken kadar.
Bu nedenle, bazal hücreli nevus sendromu gelişme riski olan çocukların yaşamlarının ilk yıllarında medullablastom varlığı düşünmek çok önemlidir.
Hastalar sendromu olmayan hastalara göre daha iyi bir sağkalım oranı var.
Tedavi:
Radyoterapi ve / veya kemoterapi tedavisi ile desteklenmiş cerrahi rezeksiyon genellikle medülloblastomlar ortadan kaldırmak için önerilmiştir. Gorlin sendromu durumunda, bu tümörlerin başlangıç yaşı daha erken olduğu; altın artı radyoterapi uygulanan, (örneğin büyüme bozuklukları ya da endokrin disfonksiyon gibi) uzun süreli sekel er riski önemlidir. Buna ek olarak, ışınlanmış alanlarda, BCC veya meningiomada gelişme riski büyük ölçüde artar. Bunlar bazal hücreli karsinom çok daha agresif ve dirençli tedavi olarak kanıtlamak.
Bu nedenlerden dolayı, sendromu enfekte kişilerin radyasyon terapisi ile tedavisi mümkün olduğunca kaçınılmalıdır.
Medulloblastomlardır da 42 kemoterapi tedavileri karşılamak, bu tedavi yöntemi, en azından genç hastalarda tercih edilmelidir.
Bu tür bazı mental retardasyon veya falx bir kalsifikasyonlar olarak, erken yaşta bulunan Gorlin sendromu, özellikleri, tümörün yol açtığı belirtiler sadece yorumlanabilir. Buna ek olarak, örneğin deri lezyonları ya da üst çene keratokistlerin Hastalığın en klasik işaretler, ancak daha sonra görüntülenir.
Bu nedenlerden dolayı, bazal hücreli nevus sendromu tümör varlığına rağmen bu yaşta tespit etmek zor olabilir.
Bazı yazarlar kemik değişiklikler, erken işaretleri ile, bir parçası olduğu için medulloblastoma hastalığın en önemli kriterlerinden biri haline gerektiğini düşünüyorum. Genetik araştırmalar ve titiz klinik muayeneler 3 yaş ve aileleri önce olası bir Gorlin sendromu arama, Medulloblastomlu tüm hastalara sunulmalıdır.
Medulloblastom en sık posterior çocuklarda fossa habis bir beyin tümörü olduğunu. Bu, beyin tümörlerinin yaklaşık% 20'sini temsil yetişkin beyin tümörlerinin% 1'den daha az ise.
Onun prevalansı 1-9 / 100 000 olarak tahmin ediliyor.
keşif yaş ortalaması 5 yıldır. Çoğunlukla çocuklar etkiler.
Medulloblastomlardır vermis gider ve 4. ventrikül ağırlıklı gelişir.
RİSK FAKTÖRLERİ
ÇEVRE FAKTÖRLERİ
bir faktör tespit edilmiştir. Bu barbitüratlar bir prenatal maruziyet olacaktır. Ancak, bu varsayım tüm çalışmalar ile teyit edilmemiştir. GENETİK FAKTÖRLER Gerçekler ... medulloblastom Genetik yatkınlık vakaların yaklaşık% 10'unda görülmektedir. Tümör 4 yaşından önce kendini Eğer kalıtsal bir yatkınlık olasılığı güçlüdür. Bu yatkınlıkları başlıca gelir:
genetik değişiklikler profili hala tam olarak bilinmemektedir, belirli sinyal iletim yollarının aktivasyonu tanınan prognostik faktör olmasına rağmen.
Modern teknikler medullablastom üçte göstermiştir ki, hastalık varlığı ile ilişkili bulunmuştur Bir kromozom anomalisi, izokromozom 17Q. Bu anomali kötü prognoz ile ilişkili bazı yazarlar için vardır.Benzer şekilde, büyük hücre medulloblastoma ve gen desmoplastik amplifikasyon varlığı C-Myc kötü prognoz ile ilişkilidir.
Belirtiler
Hastalığın ilk bulgusu en sık sabah özellikle baş ağrısı ve kusma intrakranial hipertansiyon neden vardır.Bu belirtiler beyinde bir hidrosefali aşırı beyin omurilik omurilik sıvısı (BOS) varlığından kaynaklanmaktadır. Aşağıdaki belirtiler de karşılaşılabilir:
Böyle diplopi (çift görme) aralıklı veya sürekli olarak görme problemleri,
Yazma problemleri, okul performansında düşüş
İlgisizlik, kayıtsızlık
semptom ve tanı, 2 ila 5 ay, başlangıcı arasındaki zaman çocukluk çağı kanserlerinin bağlamında uzun biridir ve bu belirtilerin özgüllüğü eksikliği ve onların tahsis açıklanabilir Psikolojik nedenler.
TANI
YOL? Bu tanıyı koyabilmek için temel bir sınav olduğunu MR var. Bu sinir sisteminde metastaz bir hastalık olduğu için, aşama içeren gerçekleştirilir:
Omurilik tam bir MRI
Kesin bir olası rezidüel tümörün varlığını incelemek için ameliyat sonrası MRI
10 gün operasyon sonrası BOS tümör hücrelerinin Arama
STAGING T ve M değerlerini teriminin bilanço, hastalığın aşaması belirtilebilir. Dört değer tümör için nispeten eski bir sınıflandırma, ayırt edilirler "T"
T1 <3 cm
T2> 3 cm
T3: T3a = Dördüncü serebral ventrikül ulaştı; T3b = IV ventrikül veya beyin sapı zemin ulaştı
T4 III ventrikül ve / veya omuriliği ulaşan =
Metastaz "M" için dört durumları belirlenmiştir İçin
M0 hiçbir metastaz =
BOS tümör hücrelerinin M1 = varlığı
M2 = metastaz (ler) Serebral (ler)
M3 = metastaz (lar) medüller (ler)
M4 = metastaz (ler) Sistemik (ler)
Risk grupları yorum iki grupta birinde Medulloblastom sınıflandırır:
standart risk grubu, T1, T2, M0 karşılık gelir ve tümörün tam rezeksiyonu ile tedavi edilecek
yüksek risk grubu, diğer durumlarda karşılık
HASTALIK ŞEKİLLERİ
Çoğu medulloblastomlar beyincik geliştirmek; Ancak, onlar da beynin diğer bölgelerinde ortaya çıkabilir.
WHO sınıflandırması beş ana histolojik tipleri ayırt:
Sözde klasik formu, vakaların% 70'i
dezmoplastik formu, daha iyi prognoz, vakaların% 15
Form geniş nodülarite
anaplastik formu
söz konusu bir şekilde büyük hücreli
Bir biyomoleküler sınıflandırma şimdi var:
WNT aktivasyonu, vakaların% 10 ile
Sinyal yolu SHH'nin aktivasyonu ile (Sonic kirpi) tanelerinin nöral öncüleri çoğalması üzerinde önemli bir kontrol uygular, vakaların% 30
Amplifikasyonu ile MYC (E Thompson veya 3) arasında,% 25
17Q kazanç (Thomson A ve C ya da 4) olguların% 35 ile
TEDAVİ
Hastalığın prognozu büyük ölçüde son yirmi yılda geliştirdi. Şu anda, orada standart risk medulloblastomda kadar% 85 oranında uzun süreli sağkalım oranlarında önemli bir artış. Olan
cerrahi teknikler, radyasyon tedavisi Gelişmeler ve kemoterapi gelişimi daha iyi yönetimine katkı Her iki sekel daha prognoz açısından.
tedavinin yönetimi multidisipliner ve beyin cerrahı, onkolog ve radyasyon terapisti olmak üzere pediatrik nöro-onkoloji içerir. Tedavi tedavi modaliteleri çeşitli kullanır:
Ya da stereotaktik Nöroşirürji geleneksel
Tümör alanının ışınlanması ve merkezi sinir sistemi dahil olmak üzere Adjuvan radyoterapi
metastatik için kemoterapi
Daha yakın zamanlarda proton tedavisinin daha etkili görünüyor ve daha az sekel neden
B / Diğer nörolojik belirtiler
Meningiomlar:
Meningiomada ilişkili bazal naevomatoses sadece 10 olgu literatürde bildirilmiştir. Hastaların yaşı 18 ila 64 yıl arasındadır. Cinsel hakimiyeti yoktur. Bu tümörler, önceki bir medulloblastom tedavisinin neden olduğu radyasyon bölgelerinde yetişen görünmektedir.
Zeka geriliği:
Mental retardasyon ve öğrenme güçlüğü vakaları da hastaların yaklaşık% 5'inde bildirilmiştir.
5 / DİĞER GENEL ETKİNLİKLER
A / over myom:
Gorlin sendromu etkilenen kızlar ve kadınlar yumurtalık myom sunabilirsiniz. myom en sık yumurtalık iyi huylu tümörler ve tüm over tümörlerinin yaklaşık% 20'sini temsil etmektedir. 1964 yılında onların ilk raporunda, Gorlin ve Goltz over myom Gorlin sendromu yaşayan kadınların% 75'inde mevcut olduğunu tahmin ediyorlar.
Yürütülen çalışmalar bir dizi son zamanlarda 14 ila% 24 oranında bir yaygınlık gösterdi. Ulaşmak ortalama yaşı 30.6 yaş, fakat 16-45 yıl arasında değişebilir. Bu tümörlerin yaklaşık 3,5 yıl daha genç hastalarda bulundu Ancak unutmayın. Bu nedenle, hastalar sendromu 13 yaşından önce ilk ultrason geçmesi ve düzenli jinekolojik bakım olmalıdır. Yumurtalık myomlar yavaş yavaş büyür ve 6 cm çapında bir ortalama boyutu olsa bile bazı çapı 30 cm kadar çıkabiliyor.
Bu Gorlin sendromunda karşılaşılan over myom daha sık genel nüfusa göre ikili ve kalsifiye olduğu rapor edilmiştir. Vakaların çoğunda, bu tümörler tamamen asemptomatiktir. Onlar doğurganlık karışmaz ve hastalar nadiren malign dönüşüm geçirmek. Bu tümörlerin tedavisi cerrahidir.
B / kalp Myomlar:
Kalp myom vakaları bazal hücreli nevus sendromu ile bağlantılı olarak bildirilen ancak nadir oylandı. Bunlar, bir kalp fibroma 3 tüm hastaların% 5 bulunmaktadır. Bunlar kalp tümörlerinin sadece% 5'ini oluşturuyor. Bunlar çocuklukta ve 10 yaşın altındaki çocuklarda vakaların% 85'inde görülür. Onların en sevdiğim nokta interventriküler septum olduğunu. Onlar yaşamın çok erken dönemlerinde ortaya çıkar ve hastanın erken ölüme kadar yol açabilir çünkü bu tümörler hızla rastlanmaktadır.
Erkeklerde C / hipogonadizm:
Gorlin sendromu olan erkeklerde çok nadiren hipogonadizm sunabilirsiniz. Hipogonadizm gonadlar yetersiz iç salgı olduğu bir öznenin durumdur.
6 / ETKİNLİKLER ODONTOSTOMATOLOGIQUES
A. odontojenik keratokistler
1. Genel
1956 yılında Philipsen ilk kez açıklanan karakteristik bir tabaka ile, Dünya Sağlık Örgütü ya da DSÖ, "benign tek veya çok hücreli, intraosseöz odontojenik kökenlerine göre, bir odontojenik keratokist olduğunu saldırgan davranışlar ve infiltre için katlı yassı epitel parakératinisé ve potansiyel. " DSÖ ayrıca bu iyi şekliyle "tümör" önerir neoplastik (saldırgan davranışlar, büyük genişleme, nüks eğilimi ...) doğasını yansıtır. dönem ilkel kist uzun karışıklığı sürdürmek, dönem keratokist bir eşanlamlısı olarak kullanılmıştır. Hepsi keratinizasyon yok çünkü Aslında, tüm ilkel kistler keratokistler değildir. Tersine, tüm keratokistler bugüne kadar her zaman bir dişin yerine geliştirmek yok gibi temel kistler değildir.
vadeli epidermoid kist arada eşanlamlı olduğunu. Onlar çenelerin tüm kistlerin yaklaşık% 11 temsil etmektedir. Onlar radiküler ve foliküler kistler sonra üçüncü sıradayız. Onlar maksilla daha mandibulada daha sık üç kez görünür.
kümülatif frekans normal popülasyona on kırk yıl arasında gözlenmesine rağmen odontojenik keratokistler, bütün yaş gruplarında meydana gelir. Muhafazakarlar tarafından, hastaların Gorlin sendromunda, tepe frekans önceki ve yaşamın ilk on yılında nedenle. Bu kistler, böylece hastalığın erken semptomu oluşturur. Bu bazal hücreli nevus sendromu erken tanısında dişhekiminin seçimi yerini teyit etmektedir. Onlar standart radyografi sırasında tesadüfen keşfedilen çoğu zaman. asemptomatik hastaların yüzdesi, 34 ile% 50, çalışmaya bağlı olarak değişir. Bunlar aynı zamanda, genellikle şişme, ağrı semptomlarının görünüşü ile ortaya çıkarılabilir; ama trismus, selülit, diğerleri arasında gecikmeli patlama veya diş malpozisyon ...
Bütün bu belirtiler keratokist özgü değildir unutmayın. genç bir hastada keratokist Odontojenik varlığı veya tekrarlayan veya multipl keratokistin görünümü uygulayıcısı uyarmak ve Gorlin sendromunun olası bir tanı doğru olmalıdır. Keratokistlerin yaklaşık% 5'i bu sendrom ile ilişkilidir. Bunlar, genel nüfusun sadece% 5-7 mevcut olduğunda Bunlar Gorlin sendromu olan hastalarda% 80 oranında bulunurlar. Keratokist etyolojisini açıklamak için çeşitli teoriler vardır.
Bazı odontojenik keratokistler diş plakaları gelişecektir.
Nitekim, diş mikropların oluşumunu izleyen, diş şeritler her iki çenede de çözülür. Bu fenomenin sırasında, adacıklar ve epitel hücre kümeleri bağ dokusunda devam edebilir. Bu yapılmamış aktivasyon üzerine bilinmeyen faktörler keratokistlerin oluşum nedeni bu artıkları, epitel olacaktır. Diğer göre, mine organı veya oral mukozanın epitel bazal tabakanın Stellat retikulum kaynaklanmaktadır. Nitekim, bu diğer teori bazal tabakanın yansımaları keratokistlerin oluşumunun nedeni olduğunu varsayar. Bu teoriyi destekleyen bir argüman olduğunu, bu kistlerin gözlenen nüksler bile etkilenen kemik segmentlerinin rezeksiyonu. Bu tekrarlama kökeni bu kemiğin dışında ve muhtemelen komşu yumuşak dokuda olduğunu varsayar. Bu oral mukoza gibi diş lamina hem ektodermal dokuların sorumlu olduklarından, bu iki teori uyumsuz olmadığı unutulmamalıdır.
2. Özellikler
çene lezyonlarına histolojik tanı bunun histolopathologique analizi ile yapılabilir. Radyolojik incelemede kesinlikle uygulayıcısı rehberlik ancak hiçbir durumda kesin tanı yapabilirsiniz. Bu sınav içerik analizi oluşmaktadır ve kistik duvarlar kistin tam rezeksiyon sonrası ya da onun marsupiyalizasyon drenajı sırasında bunun bir parçasını aldıktan sonra elde edilir. En yazarlar tarafından listelenen temel histolojik özellikleri 1966 yılında Pinborg ve Hansen olanlardır:
- Az ya da hiç damak tadınızı ve güçlü bir mitoz ile kalınlıkta genellikle çok ince ve düzgün skuamöz epitel astar;
- Bir iyi tanımlanmış bazal hücre tabakası. Bunu oluşturan hücreler küp veya silindirik şekiller ve bir çit düzenlemesi en sık düzenlenmiş;
- Sıklıkla hücre içi ödem gösteren dikenli hücre dört ile sekiz hücre tabakalarından ince bir tabaka;
- Keratinizasyon genellikle parakeratotik (çekirdeklerin varlığı) 'dir. Bu keratokistlerin 83-97% 'unda görülür. Bazen bir keratinizasyon orthokeratosic tür (hayır çekirdekleri) ya da para- ve orthokeratosic hem karşılamak mümkündür. Daha az agresif ve parakeratotik tür olarak daha az tekrarlayan olurdu çünkü Martin gibi bazı yazarlar için, orthokeratosic tür kişiye özel olmalıdır;
- Keratin tabaka genellikle dalgalı olduğunu. Sadece keratinizasyon diğer odontojenik kistler keratin üretebilir çünkü belirli bir kriter keratokist değildir.
- Lifli kist duvarı genellikle ince ve genellikle değil inflamasyon;
- Genellikle epitel adaları veya kistler "kız" izole çevre içeren ince bir kabuk konjonktiva. Kistin luminal içeriği kalın, beyaz, soluk veya sarı krem bir sıvı yönü vardır. Keratin bir varlık, ancak değişen miktarlarda bulunmaktadır. Enflamatuar reaksiyon durumunda, bu içerik kolesterol kristalleri (parlak pullar biçiminde olmak üzere), beden hiyalin, polinükleer ve bakteri kümeleri bulunur. Kist duvarı iltihaplı Ek olarak, epitel kalınlaşmış ve yavaş yavaş o odontojenik inflamatuar sıradan kistlerin oldukça benzer bir keratinize olmayan epitelde dönüşür. FNA da çene lezyonlarına tanısında önemli bir testtir.
Aslında, FNA bu keratin varlığı tanı yardım öğesidir (ama yalnız o kesin tanı koymak için yeterli değildir, çünkü böyle dentigeröz kistleri, primer kök veya bir epitel sahip diğer kistler keratinizedin edilebilir yüzey).
Yukarıda tarif edildiği gibi, ancak, enflamatuar reaksiyon varlığı bazen de doğru sitolojik tanı önleyebilir ve böylece keratokistlerin parakératinisation progresif kaybı kaynaklı vb. Bir teşhis keratokist sevk için İİAB diğer öğelerinden biri kist protein içeriğinin tahminidir.
Bu içerik, sıvı, 100 ml başına proteinin en az 4 gram ölçüldüğünde, tanısal keratokist daha sonra nispeten güvenlidir. Bu ölçüm için, inflamasyon varlığı da kötü bir etkisi vardır.
Gerçekten de, bu protein içeriğini artırmak ve dolayısıyla yanlış negatif oluşturulması katılmıştır eğilimindedir. Cerrahi dönmeden önce tanı gerçekleştirmek için yöntemler de vardır. Bu teknikler henüz yaygın bir uygulama değildir. İlk kist sıvısında KAC antijen (keratokist antijen) aramasıdır. Bu varlığı tanıyı doğrular. İkinci sitokeratin 10 immünohistokimyasal ifade belirlenmesidir.
Nitekim, bu keratinize epitel bir belirtecidir. Bu, aynı zamanda sinir kökü kistleri 10 ve% dentigeröz kist% 3 oranında, keratokist yaklaşık% 50 olarak bulunmuştur. Ancak bu lezyonlar olarak anlayışlı keratokist olmaz bu sitokeratin ifadesi çalışma dekompresyon sonra ifadesini kaybedecek gibi görünüyor.
3. Klinik ve radyolojik özellikleri
Yer:
daha sık maksillada mandibulada keratokist odontojenik koltuk. Nitekim, ilk bölgedeki yerleri ve ikinci molar dişler ve daha ön yerlere izledi özellikle açı düzeyi ve ramus de, mandibulada daha sık üç kez oluşur.
Maksiller keratokist merkezinde biraz daha erken prémolomolaire bölge ve köpek ve son derece ön bölgede yer almaktadır.
Başlangıç yaşı: Gorlin sendromu ile ilişkili odontojenik keratokistler yaşamın ilk on yılında, genel olarak, geliştirir ve bu sendromu ve keratokist ile ilişkili olmayan, aksine, 20 ve 30 yaş arasındaki pik insidansı ulaşmak dördüncü on (yaş ortalaması 37 yıl) boyunca, daha sonra keşfedilir.
Çünkü bu erken gelişim, hastalığın ilk göze çarpan belirtiler birini oluşturmaktadır ve bu sendromun tanısında diş hekimliği için tercih yeri onaylayın.
Boyutu:
Bunların hacmi yan-alt çeneye (18) içindeki bir su baskını birkaç milimetre arasında değişen, daha çok değişkendir. Genellikle rutin röntgen inceleme sırasında, daha sonra keşfedilen, çünkü onlar zaten önemli bir hacme ulaşmıştır olması nadir değildir. 5 santimetrelik ortalama uzunlukta. (34)
Cinsiyet oranı:
Normal popülasyon Erkeklerde daha sık 2/1 bir cinsiyet oranı kadınlara göre daha etkilenir. Bazal hücreli nevus sendromu olan hastalarda eksilerini tarafından, cinsiyetler arasındaki fark yok ve hatta kadınların biraz daha sık erkeklere oranla etkilenir gibi görünüyor. (5) Semptomlar: keratokist çoğunluğu herhangi bir belirti ücretsizdir. Nitekim,% 50 34 asemptomatik ve böylece diş ya da radyolojik inceleme yordamı sırasında sadece keşfetti vardır. Belirtiler var, onlar acı verici olabilir, bir kemer ya da ilerici kemik şişmesi görünümünü tarafından en sık ortaya çıkar.
enflamatuar fenomeni daha sonra seyrek değildir ve apseler, selülit ve hatta bazen eşlik fistulisations bir tetanos meydana gelmesi ile karakterize edilir. Bu olayların dışında, keratokist çünkü kaçıklıkların, diş malokluzyonların veya gecikmelerden da kızarıklık neden olabilir ve uygulayıcısı meydan hangi tespit edilebilir.
Tüm bu bulgular hiç de belirli keratokist değildir ve radyolojik muayene ve tanıyı doğrulamak için özellikle histopatolojik siparişi gerektirecektir unutulmamalıdır.
Büyüme:
Keratokist ana klinik özelliklerinden biri güçlü büyüme potansiyeli olduğunu. Bu hatta onları benign tümörleri dikkate WHO açan kriterlerden biridir. Çeşitli teoriler bu özelliğini açıklamak için öne sürülmüştür:
- Bir ilk açıklama keratokist içinde kollajenaz enzimin varlığı olacaktır. O bu güçlü büyüme potansiyeli açıklayan kolajenolitik aktiviteden sorumlu olacaktır;
- Başka bir teoriye göre kist tarafından salgılanan prostaglandin E ve F tarafından uyarılan kemik erimesinin varlığıdır;
- Son olarak, kistik sıvı ve kist duvarında artmış mitotik gücünün daha ozmolaritesi de bu güçlü büyüme için sorumlu olabilir. Bu kistler uzatılması kortikal ve periost bölgelere yönelik diğer kistler gibi yana doğru uzanan ziyade süngerimsi kemik ikame kemik içindedir.
Keratokistler hantal olduğunda bazen delikli, inceltilmiş sonra edilirse, bunlar kortikal 53 (genellikle dış) kemik ve dişlerin, kenarlarında kalıplanırlar. Mandibular ve / veya majör yüz deformiteleri kırık vakaları bildirilmiştir ama ancak çok nadirdir oylandı. Böyle inferior alveolar kanalın nörovasküler demet olarak soylu unsurlar da, her zaman saygı, ama geri döndü (ve çok nadiren Paraestezyaya neden) olabilir.
Nüks, nüks mekanizması :
odontojenik keratokistler da çok yüksek nüks oranı var. Bu ana klinik özelliklerden biridir. Bu oran bazal hücreli nevus sendromu olan hastalarda daha yüksektir ve sağlıklı hastalarda (33) 'de% 25-60 civarında karşısında% 82 olduğunu. Suç işleme bu yüksek oran nedenlerini açıklayan üç mekanizma vardır:
- Kist nedeniyle veya çok ince ve gevrek kist duvarının çevrelerine Eksik eksizyonu ya komşu yumuşak dokuya kist yapışması ile kortikal kemiğin bir delinmesi nedeniyle;
- Uydu kist, unutulmuş ya da ameliyat sonrası bir mikrokist odontojenik kalıntılarından çene lezyonlarına oluşumu;
- Özgün lezyon ile hiçbir bağlantısı olan bir de novo oluşumu ise bitişik bir alanda ve hangi yeni keratokist Odontojenik gelişimi bir nüks olarak yorumlanır.Diğer yazarlara göre, nüks oranı da etkilenir:
- Boyutu, konum (Ramus), tedavi daha zor hale lezyon (özellikle multiloculaire) ve cerrahi morfolojisi
- Cerrahi teknik seçimi (konservatif veya agresif) En nüks ilk destek 5-7 yıl içinde bulunurlar.
Ancak olgular 10 yıldan fazla tedavi sonrası rapor edilmiştir.Tekrarlayan vakaların% 25'i 9 yaş ve üzeri arasında yer almaktadır.Odontojenik keratokistler tedavinin 54 önemli bir yönü (on yıl erken bir aşamada hemen hemen tüm nüks teşhis edebilmek için ideal görünüyor) uzun vadede izlenir.; Bu kistler lokal invaziv, çok yıkıcı ve yüksek nüks oranı: İlk davranış: - keratokist bazı özellikleri tümörleri veya benign ziyade kistler olarak sokulması için DSÖ getirdi- Sonra onların histopatoloji: çalışmalar göstermiştir bağ dokusu bazal tabaka tomurcukları söyledi.DSÖ ayrıca mitoz rakamları sıkça üstü bazal tabakada bulunan kaydetti;epitel bir mitotik indeks diğer çene kistlerinin göre (ama ameloblastoma benzer) yükselmiştir keratokistin;- Genetik bitirmek için: ptch gen kromozom 9q22.3-Q31 üzerinde bulunan bir tümör baskılayıcı geni olduğunu.Bu gen, bazal hücreli nevus sendromu, ancak izole edilen keratokist oluşumu de sorumludur hem de sorumludur.Ligand SHH için (düzeltilir) onkogen SMO (sonic kirpi) ile sinyal alıcısı oluşturan bir transmembran proteini Normalde ptch kodları.Farklı doku ve çoğalmasının büyüme sinyalinin bir inhibisyon olduğunda SHH reseptörünün bu harekete geçirir.Aynı zamanda, lipid metabolizmasının düzenlenmesinde rol oynar.Bir işletim ptch kaybederse eksilerini, biz bir hücre çoğalma etkisi (ve kistler ve tümörler dolayısıyla gelişimini) sahip olacaktır.
Standart radyolojik yönü:
Çok sık asemptomatik, çoğu keratokistler standart radyolojik muayene sırasında tespit edilir.Hiçbir durumda bu sınav kesin tanı yapmak.Bu tanı bir oryantasyon sadece histopatolojik analizi ile teyit edilebilir verir.çene lezyonlarına en yaygın X-ışını görünüm mono (genellikle) ya da, radyolusent yuvarlak veya oval ve ince iç bölümleri ile polygéodes olduğunu.
Maksilla olarak, sinüs veya burun boşlukları ile bindirmeleri bu özelliklerin belirlenmesi çok daha zor hale getirebilir.Keratokist çok geniş incelme ve kortikal kemik deformasyonu mümkün olduğunda.Herhangi bir hemi-maksilla üzerinde gelişebilir;kondiller her zaman saygı.
İkincisi de, çok nadiren, temas üzerinde bulunan diğer dişlerin kök rezorpsiyonu (dahil) bir unevolved bitişik diş, süpernümerari diş veya odontome sarın ve ötelemeye neden ya da olabilir.Periferik asil elementler (sinirler ...) yok ama kist geri gönderilmez. Ana ayırıcı radyolojik tanı sonra ameloblastoma, dentigeröz kistler bazı doğar.
4. Evrim
skuamöz hücreli karsinom olarak keratokist malign transformasyon vakaları çok nadirdir. Sadece 12 olgu bugüne kadar literatürde bildirilmiştir. Birden nüks lezyonun çok agresif bir davranış olarak konservatif tedaviler ve agresif tedaviler hem bir arıza olduğunda bu olasılık dikkate alınmalıdır. Bu genellikle yaşlı hastalarda görülmektedir. Vaka ameloblastik dönüşümler çok nadir de rapor fakat
. 5.. Ayırıcı Tanı
çene lezyonlarına ayırıcı tanısı çoğunlukla diğer üç kistleri yapılmalıdır:
- ameloblastoma ile:
Bu kist erkeklerde en sık görülen dördüncü ve beşinci yıllarda zirveye oranına sahiptir. Ameloblastomanın% 80'i mandibulada ve özellikle açı düzeyi ve ramus yer almaktadır. Maksiller lokalizasyonu nadirdir.
Klinik bakış açısından, ameloblastoma sınırsız büyüme ve yüksek nüks oranı ile karakterize edilir. Bu lezyonlar genellikle yeterince büyük bir hacme sahip. Genellikle asemptomatik, sadece radyografik muayene tesadüfen keşfedilen edilecektir. Aksi takdirde, bulaşıcı bir bölüm veya hareketleri ve diş anomalileri evrimle ortaya çıkabilmektedir.
Görünümünde bir radyolojik açıdan bakıldığında, ameloblastoma çoğunlukla polygéodique yönü "sabun köpüğü", düzenli sınırları ve iç bölümleri ile vardır. Geodes iyi Farklı boyutlarda, yuvarlak veya oval demarcated edilir. asil unsurları tahrip değil, bastırıldı. Sadece standart klinik ve radyolojik bulgular ile imkansız ayırıcı tanı yapma keratokist ile pek çok benzerlikler vardır. Ayırıcı tanı olacak bu nedenle ağırlıklı olarak histolojik inceleme ile değil, aynı zamanda BT ile.
- Bir dentigeröz kist ile:
Bu kist erkeklerde daha sık bir araya ikinci ve üçüncü yıllarda zirveye oranına sahiptir. Aslında üçüncü alt molar ve üst köpek ve alt ikinci premolar ilgiliydi.
Semptomlar çoğunlukla ücretsiz, dentigeröz kist bir gömülü dişin boyun yapışır ve taç çevreler. Onun keşif rutin röntgen muayenesi sırasında en sık ya da açıklanamayan diş hareketi motive edilebilir.
radyografik görünümü iyi sınırlı osteoliz, monogéodique ve çelenk veya gömülü dişin taç etrafında geliştirilen ve tamamen inşa olmasıdır. lezyon oldukça büyük olabilir ve mandibulanın artan şube istila edebilir. Bu radyolojik Gömülü dişin boyun taktıktan ve bu nedenle kerato ayırt algılamak daha zordur.
Buna ek olarak, bu lezyona komşu dişlerin kök emilmesini gözlemlemek mümkündür; hangi keratokist için daha nadirdir ve bu iki tarafın ana ayırt edici radyolojik işaretidir.
- Bir foliküler kist ile:
Bu kist kist dentigeröz bir çeşittir ancak diş köklerinin ile temas halinde geliştiren; Bu dişler eksik kurdu söylemektir. Onun keşif kalıcı dişin gecikmiş patlama motive bir grafisi vasıtasıyla ya da yapılabilir; ya (genellikle premolar bölgede) bir enfeksiyon atağı sırasında. Diğer özellikler Dentigeröz kist özdeştir.
- Banal inflamatuar kist ya da diş radiculo ile:
Bu lezyonlar en sık maksiller bulunmaktadır. Onlar tercihen ön maksilla veya mandibulanın yatay dalında erkeklerde meydana gelir. Onların zirve insidansı üçüncü ve dördüncü yıllarda arasındadır. Benzer lezyonlar daha önce görülen, radiküler kist genellikle asemptomatik ve radyolojik rutin keşif. Ancak nekrotik veya eksik işlenmiş bir çürümüş diş, yanında, şişme, irin şişkin ile karakterize bulaşıcı bölüm ortaya çıkabilmektedir. Bu kistler yavaş büyüyor ve klinik bulguların yokluğunda, önemli ölçüde genişletmek olabilir.
Bunların radyografik olarak monogéode arasında, dişe mahcup ve (inflamatuar ataklar sırasında kaybolabilir) sınır kemik yoğunlaştırma ile çevrelenmiş tepe noktası ile bağlantılı olarak, yuvarlak veya oval olmasıdır. Bunlar önemli boyutu, komşu kökleri nedeni kök rezorpsiyonu durumunda, olabilir. 59
Ayırıcı tanı ayrıca odontojenik fibroma, odontojenik miksoma, soliter kist, kemik anjiomuna veya anevrizmal kist gibi diğer kistleri inebileceği. 60 B.
B / Diğer olaylar odontostomatologiques
Diğer Diş odonto- olaylar da bazal hücreli nevus sendromu ama daha büyük bir eşitsizlik ile karşılaşılır. Nitekim, onlar her zaman mevcut değildir ve onlar zaman, kendilerini, hastalığın başlıca kriterlerden biri olan keratokist çok daha düşük bir frekans ile olduğunu. Bunlar genellikle keratokistlerin gelişimine doğrudan ilişkilidir ve onları vurgulamak (ve dolayısıyla bazal hücreli nevüs sendromu tanısı yol) için ilk çağrı işaretlerinden biri temsil edebilir
Bir / Kapanımlar veya diş malpozisyon
Bunlar ağırlıklı olarak kalıcı dişlenme ilgilidir. Nihai diş onun patlamasını önleyen kist tarafından korunur iken Süt dişlerinin bir sebat gözlemlemek mümkündür. Çalışmalar kistin zaman gelişimine bağlı olarak, ortaya koymuştur, sonuçları diş seviyesi farklıdır. Keratokist zaman kök inşaatı geliştirir Nitekim, deformasyon gibi eğrilikler olarak köklerde görülen veya süngü büker oldu.
Bir diş eleman oluşumu yan yana erken gelişirse, köklerin yırtılmasına neden olabilir. Kist diş patlama sonrası oluşan halinde eksilerini tarafından, o seyahat ve diş malpozisyon sorumlu olabilir ama her durumda kök rezorpsiyonu veya hamur mortification sorumlu olamaz.
b / Mandibular prognatizm
Olguların tahminen% 25. Bu gerçek prognatizm veya brachymaxillie bağlanmış bir pseudoprognathisme, ya da her ikisinin bir kombinasyonu da olabilir. 61 Onların desteği ortognatik cerrahi ve ortodonti aracılığıyla olacaktır. Gorlin sendromlu genç bir hastada Mandibular prognatizm (15)
c / dudak ve damak yarıkları
Yarık dudak ve / veya damak yarığı bazal hücreli nevus sendromu vakalarının yaklaşık% 5-7 bulunur. Lambrecht tarafından yapılan bir literatür Gorlin sendromu etkilenen 719 kişinin, orofasial 61 yuvası, hastaların% 8.5 olduğunu göstermektedir. Bu insanlar yuvası dudak, alveol ve damak (49 olguda) ya da tek yarık olmasına bağlı olarak iki gruba ayrılabilir (13 olgu için)
Bu malformasyonların sıklığı bazal hücreli nevus sendromu, genel nüfus ve hastalar arasında aynı değildir
.Nitekim, genel popülasyonda, dudak yarıkları, alveol ve damak insidansı olduğunu sadece 1/800 sırasıyla yarık damak karşı 1/2500 1 / 14.7 ve hastaların 1 / 55.3 Gorlin sendromu. Bu anormallik, bir yenidoğanda bir Gorlin sendromunun varlığı ilk göstergelerinden biri olabilir.
d ./ parestezi
Onlar da keratokistlerin gelişmesine bağlı ve sıkıştırır veya sinir eleman bastıran ve parestezi neden olursa kist belirgin bir boyuta ulaştığında ortaya ç. Aynı zamanda sinir yakınında sınırları ile büyük bir kistin çıkarılması bir ameliyat sonrası komplikasyon olabilir.
E / améloblastome
Birkaç vaka bildirilmiştir beri ameloblastomaların bazıları nadiren hasta Gorlin sendromunda rastlanır. Eslami Gorlin sendromu ve ameloblastoma sahip olarak elde bir 68 yaşındaki bir hastanın durumu göstermektedir.
Bu hastalıktan etkilenen hemen hemen tüm hastalar için, ilk belirtilerin erken yaşta ortaya ederken, genellikle 35 yaş öncesinde, hastanın ilk klinik belirtileri daha sonra ortaya çıkmaya başladı.
Nitekim, sadece 68 yıl 50 yaş ve çoklu keratokistler onun ilk bazal hücreli karsinom sundu. Eleman ilginç, biz Eslami İngiliz edebiyatının kaydedilen ameloblastoma ile ilişkili bazal hücreli nevus sendromu 4 olgunun 3, aynı zamanda bazal hücreli nevüs sendromu olgularının çoğunluğu daha eski bir yaşta ilk belirtileri takdim kaydetti.
Bu durumlar için yaş ortalaması 47.6 yıldır. Bu keşif daha geç bir yaşta bazal hücreli nevus sendromu olan hastalar gelişen ameloblastomanın riski daha olduğunu gösterir.
Yukarıda görüldüğü gibi, odontojenik keratokistler ve ameloblastomaların bazıları yüksek nüks oranı ve kemik yıkım büyük potansiyeli olan iyi huylu tümörleri, hem vardır.
Ancak, ameloblastoma daha da radikal cerrahi rezeksiyon gerektiren daha kalıcı ve daha infiltratif olduğunu.
Notes aux visiteurs, Notes for visitors, Note per i visitatori , Notas para los visitantes, Hinweise für Besucher, Примечания для посетителей, 遊客注意事項,Notes voor bezoekers, Ziyaretçiler için notlar
Étant seule à créer ce site , sans l'aide de quiconque , si vous voyez des oublies, ou des erreurs sur mon site, merci de m 'en informer par mail .. si vous voulez apporter des temoignages, (bien entendu de maniere anonyme, des renseignements complementaire , photos , etc , contacter moi . je me ferai un plaisir de les publier ..
.
D'autre part, pour faciliter la lecture de chacun, en langue étrangère, j'ai utilisé plusieurs couleurs d'écriture suivant les langues traduitent dans la page d'accueil . Dans les autres chapitres suivez les menus déroulants pour accéder aux traductions . :
le NOIR = francais
BLEU = anglais
VERT = italien
ESPAGNOL = violet
ALLEMAND = orange
MARRON = russe
ROUGE = chinois (traditionnel )
JAUNES FONCé = neerlandais
langues supplementaires :
turc
Utilisant google traduction , si les traductions sont erronées, merci de m'en informé également
je vous remercie d'avance .
Corinne
Beingalonein creatingthis site withoutthe help of anyone, if you seeforgotten, or errorson my site,thank youmto informby email.. ifyou want to maketestimonies, (of courseanonymousway,thesupplementaryinformation,photos,etc.,contactme.Iwould be happyto publish them.. . Moreover, to facilitate the reading of each foreign language, I have used several writing colors following languages traduitent in the home page. In the other chapters follow the drop down menus to access translations
BLACK=french BLUE =English GREEN =Italian SPANISH=purple GERMAN = orange BROWN =Russian RED =Chinese (Traditional)
Dutch = dark yellow
Using googletranslation,if the translationsare wrong, thank you alsoinformed me
I thank you in advance .
Corinne
Essere solinella creazione diquestosito, senza l'aiutodi nessuno, sesi vededimenticato, oerrori sulmio sito, graziemdi informarevia e-mail.. sesi vuole farele testimonianze, (di modoovviamenteanonima, le informazioni supplementari, foto, ecc,in contatto con me. sarei felicedi pubblicarle.. . Inoltre, per facilitare la lettura di ogni lingua straniera, ho usato diversi colori di scrittura seguenti lingue traduitent nella home page. Negli altri capitoli seguire il menu a discesa per le traduzioni di accesso
Utilizzando traduzione di Google, se le traduzioni sono sbagliate, grazie che mi haianche informato
Vi ringrazio in anticipo.
Corinne
Estar soloen la creación deeste sitiosin la ayuda denadie,si vesolvidado,oerroresen mi sitio, gracias mde informarporcorreo electrónico.. siquieres hacertestimonios,(de maneraanónimacurso,la información complementaria, fotos, etc., póngase en contacto conmigo. yo sería felizpublicarlas.. .
Por otra parte, para facilitar la lectura de cada idioma extranjero, he utilizado varios colores que escriben los siguientes idiomas traduitent en la página principal. En los otros capítulos siga los menús desplegables para traducciones de acceso
El uso de traducción de Google, si las traducciones están equivocados, graciastambién me informó
Os doy las gracias de antemano.
Corinne
Allein zu seinbei der Schaffung dieserWebsiteohne die Hilfe vonjemand, wenn Sie vergessen, oder Fehlerauf meiner Seite, ich danke Ihnen bin, um per E-Mailzu informieren..wenn SieZeugnissemachen wollen,(natürlichanonym,die Zusatzinformationen, Fotos etc. kontaktieren Sie mich. ich würdeglücklich sein, siezu veröffentlichen.. . Darüber hinaus erleichtert das Lesen der jeweiligen Fremdsprache habe ich mehrere Schreibfarben verwendet folgenden Sprachen traduitent in der Homepage. In den weiteren Kapiteln folgen Sie die Dropdown-Menüs, um den Zugriff Übersetzungen
Verwendung von Google-Übersetzung, wenn dieÜbersetzungfalsch sind,dankeichauch darüber informiert,
Ichdanke Ihnen im Voraus.
Corinne
Будучив одиночестве всоздание этого сайтабез помощикого-либо, есливы видитезабыли, илиошибки намоем сайте, спасибомсообщитьпо электронной почте.. есливы хотите, чтобыпоказания, (конечно анонимныйпуть,дополнительная информация, фотографии и т.д., свяжитесь со мной.Я был бы счастлив, чтобы опубликовать их.. ,
Кроме того, для облегчения чтения каждого иностранного языка, я использовал несколько пишущих цвета следующих языках traduitent в домашней странице. В других главах следовать выпадающее меню для доступа переводов
Alleen zijn in het creëren van deze site zonder de hulp van iedereen, als je vergeten of fouten op mijn site, dank u ben op de hoogte via e-mail .. als je wilt getuigenissen maken (uiteraard anonieme manier , de aanvullende informatie, foto's, enz., contact met mij op. ik zou blij zijn om ze te publiceren ..
.
Bovendien, om te vergemakkelijken het lezen van elke vreemde taal, heb ik verschillende kleuren te schrijven volgende talen traduitent in de home page. In de andere hoofdstukken volgen de drop down menu's om toegang vertalingen. :
BLACK = french
BLUE = Engels
GROEN = Italiaans
SPAANSE = paars
DUITSE oranje =
BROWN Russische =
ROOD = Chinees (traditioneel)
DONKER GEEL = Nederlands
Met behulp van google vertaling, als de vertalingen zijn fout, dank u ook op de hoogte me
Ik dank u bij voorbaat.
Corinne
Eğer unuttuysanız, veya sitemde hatalar bkz eğer herkes yardımı olmadan bu siteyi oluştururken yalnız olmak, (siz ifadelerini yapmak istiyorsanız .. e-posta ile bilgilendirme m elbette anonim bir şekilde teşekkür , tamamlayıcı bilgiler, fotoğraflar, vs, bana ulaşın. Ben bunları yayınlamak için mutlu olurdu ..
.
Ayrıca, her yabancı dil okuma, ben ana sayfada diller traduitent aşağıdaki birkaç yazılı renkler kullandık kolaylaştırmak için. Diğer bölümlerde erişim çeviriler için aşağı açılır menülerle izleyin. :
SİYAH = fransız
MAVİ = İngilizce
YEŞİL = İtalyan
Mor = İSPANYOL
ALMAN turuncu =
KAHVE Rus =
KIRMIZI = Çince (Geleneksel)
KOYU SARI = Hollandalı
Ek diller:
Türk
Çeviriler yanlış ise google çeviri kullanarak, siz de beni bilgilendirdi teşekkür