
Über Syndrom
Z U S A M M E N F A S S U N G
Das hat der Gorlin-Syndrom? DEFINITION
DR Gorlin RJ, Chirurg-Zahnarzt und Genetiker, seinen Kurs
Genetischen Merkmale Basal-Goltz-Syndrom
A / GENERAL
B / Gen in der Basal-Goltz-Syndrom BETEILIGT
1. Allgemeines
2. Theorie der Doppelveranstaltung
3. Transduktion Way Patched / Sonic-Hedgehog
Schauspieler Übertragungsweg gepatcht / Sonic Hedgehog
Die Übertragungsweges der gepatchten Betriebs / Sonic Hedgehog
Signalweg Patched / Igel Sonic
Arten von Mutationen und Lage
Allgemein Veranstaltungen im Detail die Basal-Goltz-Syndrom
1 / EVENTS Dermatika
A / Naevi und Basalzellkarzinome Allgemein
B / palmoplantar Hyperkeratosis Allgemein
C / Dermatologische Ereignisse
2 / SKELETON EVENTS
A / Gehirn Verkalkungen Umschläge
B / Abnormale Schädelform
C / Rippenanomalien
D / Wirbelanomalien
E / Abnormale Knochen der Hand und Fuß
F / Andere Skelettmanifestationen
3 / AUGEN EVENTS
A / Angeborene Fehlbildungen des Globus
B / Tumor Globus Prozess
4 / EVENTS NEUROLOGIC
A / Méduloblastomes Statistik
B / Andere neurologische Symptome
5 / SONSTIGE ALLGEMEINE VERANSTALTUNGEN
A / Eierstock Myomen
B / Herz Myome
C / Hypogonadismus bei Männern:
6 / EVENTS ODONTOSTOMATOLOGIQUES
Ein / das odontogene Keratozysten
B / Andere Veranstaltungen odontostomatologiques
a / Einschlüsse oder Zahnfehlstellungen
b / Progenie
c / Lippen- und Gaumenspalten
d / Parästhesien
e / Die améloblastome
Das hat der Gorlin-Syndrom? DEFINITION
Gorlin-Syndrom, auch als Basalzellkarzinom mole-Syndrom (NBC) bekannt, ist eine vererbte Erkrankung, die durch eine Reihe von Entwicklungsstörungen und einer Prädisposition für verschiedene Krebsarten.
Die Prävalenz wird auf zwischen 1/57 und 1/256 000 000 geschätzt, mit einem Verhältnis männlich / weiblich 1: 1. Klinische Manifestationen schließen die Anwesenheit von zahlreichen Basalzellkarzinome (BCC), Keratozysten der Backen, Hyperkeratose der Handflächen und Fußsohlen, Skelettanomalien, ektopische intrakraniale Verkalkungen und Gesichtsdysmorphien (Makrozephalie, Lippen-Kiefer-Gaumen und schwere Augenanomalien). Geistige Retardierung ist in 5% der Patienten beobachtet. Basalzellkarzinome (von Papeln, die die Farbe der Haut zur Behandlung von Colitis-Platten mit einem Durchmesser von 1 bis 10 mm) sind in der Regel auf dem Gesicht liegt, Rücken und Brust. Die Anzahl der Basalzellkarzinome reichte von einigen wenigen bis zu mehreren Tausend.Skelettanomalien (die die Form der Rippen, Wirbelsäule und Schädel) sind häufig. Kann auch auftreten, Auge, Harn-und Herz-Kreislauf-Erkrankungen. Unter den Patienten mit Gorlin-Syndrom, zu entwickeln 5-10% Medulloblastom, die eine wahrscheinliche Ursache des frühen Todes ist.
Das Syndrom wird durch Mutationen im Gen PTCH1 verursacht und wird autosomal-dominant mit kompletter Penetranz und variabler Expressivität vererbt. Die klinische Diagnose wird auf bestimmte Kriterien. Die Diagnose wird durch Forschungs Mutationen bestätigt. Die genetische Beratung ist obligatorisch. Eine vorgeburtliche Diagnostik ist durch Sonographie und DNA-Analyse fetaler Zellen durch Amniozentese oder Chorionzottenbiopsie möglich erhalten. Die wichtigste Differentialdiagnose umfasst Bazex Syndrom, multiple trichoépithélium und Muir-Torre-Syndrom Syndrom (siehe dort). Die Unterstützung erfordert einen multidisziplinären Ansatz.
Keratozysten durch chirurgische Resektion entfernt. Im Fall von Basaliomen, ist eine Operation angezeigt, wenn die Anzahl der Läsionen begrenzt ist. Andere mögliche Anwendungen sind Laser-Ablation, die photodynamische Therapie, und lokale Chemotherapie.
Strahlentherapie ist zu vermeiden. Die Analoga von Vitamin A kann eine präventive Rolle gegen die Entwicklung von neuen CBC zu spielen. Lebenserwartung nicht wesentlich verändert, aber Komplikationen können in erheblicher Morbidität führen. Eine regelmäßige Überwachung durch ein multidisziplinäres Team (Dermatologen, Neurologen und odontologists) ist unerlässlich. Die Patienten sollten vermeiden übermäßige Exposition gegenüber UV-Strahlen.
Publisher (n) Experte (n)
DR Gorlin RJ, CHRIRURGIEN- Zahnarzt und Genetiker, seinen Kurs

Robert James Gorlin wurde 11. Januar 1923 in Hudson, New York geboren und starb von Lymphomen 29. August 2006 in Minneapolis, Minnesota im Alter von 83. Trat in die Armee im Zweiten Weltkrieg, er studierte Zahnmedizin an der University of Washington, wo er im Jahre 1947 absolvierte er seine Ausbildung durch den Erwerb eines Master of Chemistry an der Universität Staat Iowa. Nach zahlreichen akademischen Positionen, im Jahr 1956 erhielt er eine Stelle als Professor und Direktor des Oral Pathology Abteilung an der Universität von Minnesota, wo er bis zu seinem Tod blieb.
Seine Karriere nahm eine Wende im Jahre 1940, als Gorlin entdeckt ein Buch von Helen Curth Dermatologen Umgang mit achantosis nigricans. Diese Erfahrungen helfen dabei, Gorlin in "syndromologiste". Die achantosis nigricans ist eine Hauterkrankung, die durch vegetieren papilläre Hypertrophie und Pigmentierung vor allem in den Achselhöhlen, Hals und Genital-Oberschenkel Regionen, in denen die Haut erscheint rau, verdickt und quadriert lokalisiert. Das dermatologische Bedingung hat bösartigen Formen gekennzeichnet durch eine Verbindung mit einem Adenokarzinom des Magens. Es ist eine dermatologische Erkrankung, die anderen Systeme des Körpers betrifft. Diese Entdeckung hat Gorlin, die dann gefragt, ob bestimmte systemische Erkrankungen könnte oralen Manifestationen präsentieren gefesselt. Gorlin wurde international für seine Arbeit an oralen Manifestationen vieler Krankheitsbilder bekannt. Im Laufe seiner Karriere, Robert J. Gorlin und seine Kollegen untersuchten und nannte etwa 100 Syndrome und beteiligte sich an der Isolierung und Charakterisierung von Genen, die für etwa die Hälfte von ihnen verantwortlich.
Der Name des Gorlin zu einer gebräuchlichen Bezeichnung, 7 nicht nur in der Genetik, sondern auch in der Medizin vertraut machen. Dies veranlasste Dermatologen, Genetiker und Pathologen zu Gorlin als einer von ihnen zu betrachten. Robert J. Gorlin wird ein sehr fruchtbarer Schriftsteller zu sein. Er hat über 600 Artikel, 60 Buchkapitel verfasst und Co-Autor oder bearbeitet 20 Bücher. Einige von ihnen haben sogar in Spanisch, Russisch und Japanisch übersetzt. Der Name des Gorlin hat sich besonders untrennbar mit der Basalzell-Nävus-Syndrom, auch bekannt als Gorlin-Syndrom bekannt geworden.
1960, beschreibt zum ersten Mal die Zuordnung zahlreicher Haut Basalzellkarzinome auf den ganzen Körper, einschließlich der unbelichteten Bereiche in der Sonne mit Rippen Deformitäten Knochenläsionen und Defekte und Augentumoren erscheine . Er betrachtet diese Anomalien als Teile eines einzigen Syndrom als heute unter dem Begriff Gorlin-Syndrom. Es wird in dem Verständnis und der Entdeckung der ursächliche Gen, das Gen PTCH helfen.
Im Laufe seiner Karriere an der Universität von Minnesota, führte ihn umfangreiches Wissen zu den Kursen in der Pathologie, Dermatologie, Kinderheilkunde, Geburtshilfe, Gynäkologie und otorhino Throat zu geben. Er war auch Präsident der International Association for Dental Research, der American Academy of Oral Pathologie und der Internationalen Gesellschaft für Mund-, Gesichts- Biologie. Für seinen großen Beitrag zur Wissenschaft, Medizin und Gesellschaft, hat Robert J. Gorlin viele renommierte Auszeichnungen im Laufe seiner Karriere erhielt. Robert J. Gorlin ist daher eine Ausbildung Zahnarzt, der durch seine Arbeit konnte die Medizin und Genetik sowie Zahnheilkunde zu überqueren.
Genetischen Merkmale Basal-Goltz-Syndrom
A. ALLGEMEINE
Lange bevor Gorlin RJ, dieses klinische Einheit, wie der Begriff Basalzell-Nävus-Syndrom bekannt ist, hat bereits Gegenstand Beschreibungen, unter anderen Namen. Im Jahre 1894 Jarisch
beschreibt kurz die Kombination von Basalzellkarzinom mit Skelettanomalien bei einem Patienten mit Skoliose. Dabei spielt es keine Erwähnung von anderen
Zeichen der Krankheit.
Im selben Jahr, berichtet Weiß Familiengeschichte eines Patienten mit mehreren Basalzellkarzinome, die sein ganzes Leben lang erscheinen fortgesetzt.
Im Jahr 1932 beschreibt Nomland Basalzellkarzinome aus angeborenen Pigmentmalen.
Im Jahr 1939 Straith ist der Erste, der Kieferzysten, mehrere Haut Basalzellkarzinome in drei Mitglieder einer Familie zu verbinden.
Er wartet, bis 1951, dass zwei Dermatologen, Binkley und Johnson beschreiben beide Hautkarzinome und Zysten der Kiefer und
Veränderungen im Gehirn.
Es war im Jahr 1960 in Minneapolis als Gorlin, Zahnarzt und Goltz, Dermatologen führen die erste umfassende Beschreibung dieses klinische Einheit sie ein Syndrom zu prüfen. Sie werden ihm den Namen des Basalzell-Nävus-Syndrom mit multiplen Zysten des Oberkiefers und gespaltene Rippen zu geben.
Seitdem kann das Syndrom durch verschiedene Namen wie Gorlin-Syndrom oder Gorlin-Goltz Gorlin-Goltz Phakomatosen, Basalzell-Nävus-Syndrom bezeichnet werden ...
Die frühesten bekannten Fälle von Basalzell-Nävus-Syndrom wurden in den 1960er Jahren wegen der follikulären Zysten, gespaltene Rippen und andere Stigmata der Gorlin-Syndrom ergeben, wurden auf zwei erwachsene männliche Skelette Skelette der ägyptische Sammlung gefunden das Institut für Anthropologie der Universität von Turin. Diese Skelette wurden in Assyut in Unterägypten und das Datum vom dynastischer Zeitraum exhumiert.Dynastic Frist beginnt mit der Vereinigung von Ägypten, einmal in zwei Königreiche, die Nord und Süd geteilt, und ist etwa 3000 v.Chr.
Das Basalzellkarzinom Nävus-Syndrom ist eine vererbte genetische Krankheit mit autosomal dominant. Jedoch sporadische Fälle wurden berichtet: in 60% der Fälle wird kein anderes Familienmitglied erreicht und 35-50% dieser Fälle dar néomutations. Daher das Fehlen einer Familiengeschichte schließt nicht aus, die Diagnose dieser Krankheit.Es wird durch Mutation des PTCH-Gen verursacht, in 9q22.3-q31 angeordnet, die ein Gen
Tumor-Suppressor.
Die nävoide Basalzell eine hohe Penetranz die die Häufigkeit, mit der ein Gen manifestieren ihre Wirkungen ist. Es wird bei 97% geschätzt. Dies bedeutet, dass 97% der
Individuen, die dieses Gen leiden an mindestens eine der Erscheinungsformen der Krankheit.
Die Krankheit hat auch eine variable Ausdrucks; Ausdrucks wobei die Qualität des mit variabler Begriffe quantitativ vorliegenden Gens. Die Krankheit manifestiert sich in so viele Symptome eines Patienten variabel zu einem anderen.Es gibt mehr als vierzig diagnostischen Kriterien. Allerdings glaube jeder Patient mit dieser Krankheit nicht alle Kriterien zu präsentieren. Diagnostische Kriterien wurden in zwei Kategorien, Dur und Moll, abhängig eingestuft
Deren Häufigkeit. Es ist Möglich, Eine Diagnose von Basalzell-NAVUS-Syndrom bei Patienten vor mindestens Zwei Hauptkriterien BZW. Gesellschafter machen
Ein Wichtiges Kriterium in two Nebenkriterien.
Die Prävalenz 1/56000 ist mit Einem Geschlechterverhältnis männlich / weiblich 3/1. Nach Ghailan 1/60000 ist die Männer und Frauen und Prävalenz Sind in betroffenen
Gleichen Proportionen. Ende erreicht This is the Gleichstellung Geschlechter der sehr gut mit der-Syndrom autosomal-dominant Erklärt.
Laut Wolff, sterben von Prävalenz reicht 1/60000 bis 1/120000 Beide Geschlechter gleich und Sindh Häufig betroffen. Überdies Syndroms in Einer von Gruppen vielzahl Auftritt
Kulturelle and a Nicht Scheint Vorliebe für bestimmte Art von Top-have.
Basalzell-NAVUS-Syndrom bleibt Eine seltene Erkrankung, Wie sie DERZEIT have nur in der Literatur 700 Fälle berichtet Worden. Jedoch ist der in der Ersten Zahnarzt Position, um zu sterben, machen diagnostizieren, Weil maxillofazialen Zahn Manifestationen Zählen zu den Ersten Manifestationen Waren dieser Krankheit.
B. Basal-Gen in der Goltz-Syndrom beteiligt
1. Allgemeines
Die für Basalzell-NAVUS-Syndrom der PTCH Menschen Würde Zum Ersten Verantwortliche Gen Mal im Jahr 1996 von Zwei Forschungsteams entdeckt. Seine Entdeckung ist neu Ziemlich
genetischen Mechanismen alle sterben für sterben nicht gut Erkrankung verstanden Wird von der Wissenschaft, bleiben einige Elemente unklar.
This hotel Liegt Auf dem Arm Langen von und genauer Chromosom 9 in 9q22.3, Wird of this Gen Gefunden bei Patienten mit-Gorlin-Syndrom, Sondern Auch in den Gefallenen, mutiert
sporadischer Basalzellkarzinome.
Of this Gen spielt Eine Rolle in der embryonalen Entwicklung Wichtige und der Kontrolle der Zellproliferation.
In Einem Artikel aus dem Jahr 1982 offers Eine ganz andere Fitzpatrick Theory Daten der Aktuellen, um den Ursprung of this Syndrom erklären. Wissenschaftler Die Zeit der Ihre Techniken have in der Tat sterben Könnte Genetische Anomalie, sterben wir heute kennen, zu Erkennen. Er Sagte , Multi-Die systemischen Anomalien Könnte sterben Einer Existenz enzymatischen Defekts Nicht erklären. Das Vorhandensein Eines solchen Mangels der während der Ersten Trimester Schwangerschaft und can sterben Skelettanomalien Verkalkungen Umschläge das Gehirn zu erklären.
Später Könnte Eine Abnormalität in DEM enzymatischen Mechanismus Steuerung der Bildung von zur Hoch Naevi, Basalzellkarzinome, palmoplantare und Gruben Keratozysten Führen. Aber this Theorie ist unzureichend, Weil sie um den Ursprung schweigen permetpas Tumoren Wie der neurologischen Medulloblastom verstehen.
2. Theorie der Doppelveranstaltung
Die Studien Tumoren in Basalzell-mole-Syndrom ergab das Vorliegen von Einem verlust der Agentur für heterozygosity 9q Region wurde nahe LEGT, sterben Dass PTCH Gen Könnte Ein Tumor-Suppressor-Gen Brust. This Begriff von Anti Onkogen, Dessen Archetyp ist das Retinoblastomgen (Rb) , ist of this Auf dem verlust Verband beider Allele Menschen in EINEN Tumor (verlust der Heterozygotie 13) von der Durch Knudson vorgeschlagene Modell basiert 1971. Nach Wadenfänger "Theorie der Beiden Mutationsereignisse" sterben für sterben erste Mutation Fehlbildung Syndrom Verantwortlich prädisponieren, um das Auftreten von Tumoren Zweite Würde und sterben zum Auftreten von Tumoren Führen.
Nach Cohen, so that Ein Tumor-Suppressor-Gen inaktiviert Sind Zwei Phänomene erforderlich Wir. Die Erste ist sterben Übertragung Nach Eines Allele. Das ist der Zweite Phänomen verlust other von Allelen, Da der verlust der Heterozygotie (LOH Oder) Bekannt. WENN Allel Sind Beide verloren, Tritt Tumoren. Ein verlust der Heterozygotie Wurde für Basalzellkarzinome, Keratozysten und Medulloblastom, Die Drei der WICHTIGSTEN Merkmale Sind demonstriert Gorlin-Syndrom.
In der Formen Familiaren-Gorlin-Syndrom, so Wird sterben bei der erste Mutation kann sein und Wird Empfängnis vorhanden in allen Körperzellen gefunden. This erste Mutationsereignis, erzeugen Einer für Prädisposition Basalzell-NAVUS-Syndrom bei den Nachkommen zu Entwickeln, ist in Dominanz übertragen. of this Ereignis und bleibt latent macht nur that,, ideal für Eine Zweite Zwischenfall Auf dem Chromosom
homologiert gleichzeitig Locus wurde zu DEM Auftreten-Gorlin-Syndrom. In Familiaren Gefallenen, der Durcheinander nur EINEN Einzelne einzigen Mutationsschritt (Das Erste seit Wadenfänger genetischen Materials vererbt). Durch gegen das Auftreten-Gorlin-Syndrom in Einem Individuum ohne Familiengeschichte
Erfordert das von Zwei Auftreten aufeinanderfolgenden Mutationsereignisse in demselben Individuum.
3. Transduktion Way Patched / Sonic-Hedgehog
PTCH Gen humane das von Patched Homolog-Gen in Drosophila. Gen show this is a Segmentierung Drosophila-Gens (oder POLARITÄT) in der Bahn der auftretenden
Transduktion gepatchten / Sonic Hedgehog, sterben bei der Kontrolle der Entwicklung und der embryonalen Zellproliferation beteiligt ist. Sind fungiert Regulator der Zellzyklus als der Durch anhalten Zellteilung in Abwesenheit von Liganden und aktivierung, sobald sterben verbindung with the Liganden Hergestellt.
Gorlin-Syndrom verursacht Schaden am Gehirn, Rippen, Wirbeln, Mitglieder ... im Wird Diesen Geweben exprimiert Patched.
Schauspieler Übertragungsweg gepatcht / Sonic Hedgehog:
Obwohl große Letzten jahren Fortschritte in den beim Deschamps des Betriebs der Signalwegs gepatchten / sonic hedgehog Hergestellt gerechnet wurden, Sind viele
Unsicherheiten Müssen noch in der Zukunft zu klären.
- Das Protein Patched Phenylthiocarbamid oder Durch Die PTCH Gen kodiert Glykoprotein is a, das aus 1447 Aminosäuren mit Zwölf Transmembrandomänen,
Zwei große extrazelluläre Schlaufen für sterben Bindung Liganden der SHH, weiten intrazellulären Schleife einen, Einer intrazellulären aminoterminalen Domäne und Einer carboxyterminalen Als Auch notwendig intrazellulären Domäne.
Es zeigt Sich in Basalzell-NAVUS-Syndrom mutiert ist, Sondern Auch in Basalzellkarzinome seltene Fälle von und Medulloblastom.
Die meisten Mutationen Sind Nonsense-Mutationen, sterben in der Synthese Eines verkürzten Proteine Führen.(Lacombe) Gene Patched 1
. - Die-Protein Sonic Hedgehog (SHH), das Durch das Gen in 7q36 shh befindet kodiert of this 45-kDa-Protein in two Wird Domänen gespalten: eine Amino-terminale 20 kDa Zn Domäne von mit Einer aktivität und Einer Hydrolase-Carboxy-Terminalen Domäne von 25 kDa autokatalytisch setzen dotiert
Cholesterin-Transferase-aktivität.
Die Rolle des Cholesterins in diesem Signalweg ist nicht bekannt Vollständig, Aber es Könnte Eine Rolle bei der Beschränkung von der Ausbreitung SHH-Molekül spielen und verteilung
Raumwirkung.
- Das wird schließlich Smoothened-Protein (BBS), das Durch das Gen in DEM 7q31-32 Smoothened befindet kodiert.
Es Ist Ein Protein von 115 kDa, 787 Aminosäuren aus der Familie der Serpentinen Rezeptoren Jahr G-Protein gekoppelt Sindh (Die Eine zur Familie der Homologie Wnt-Rezeptoren zeigt) angehören, zusammen. Letzterer hat Eine extrazelluläre Domäne aminoterminalen, Carboxy Eine intrazelluläre Domäne und Zwei bereiche (Die Dritte intrazelluläre Schleife und sterben siebte Transmembran-Domäne) Signalübertragung im beteiligt.
Die Übertragungsweges der gepatchten Betriebs / Sonic Hedgehog:
Gepatcht is a für Rezeptor SHH Liganden Rolle. In Abwesenheit von SHH, SMO Patched and a Bilden inaktiven Komplex. gepatcht Wirkt als Ein
negativer Regulator, der Ein Inhibitor SMO.
WENN SHH binden Jahr Patched kommt, die ist letztere Dann ist frei inaktiviert und SMO, um das Signal Innerhalb der Zelle zu übertragen.
Verschiedene Mechanismen Worden Sind vorgeschlagen, äh sterben aktivierung SMO und zu Signaltransduktion erklären:
-. In Einer Ersten Hypothese, geflickt and a Smo Komplex Bilden Bindungsliganden SHH-Jahres Plan Ergebnis zu Seinen Rezeptor Patched Einer der Konformationsänderung letzteren und DAMIT sterben von Dissoziation Patched / SMO-Komplex. ASM Dann Wäre frei, um das Signal Innerhalb der Zelle zu übertragen .
- In jüngerer Zeit schlägt eine zweite Hypothese, eine nicht-stöchiometrische Regelung durch Patched. Tatsächlich zeigten Versuche, dass die Bindung von SHH ein Ergebnis diminutionde Patched und erhöhte GOS in der Zellmembran. Das Verhältnis zwischen
atched und SMO würde nicht stöchiometrischen, dh, die regeln, wäre die Patched 16 sein
SMO Veränderung, sondern eine reale Anlage mit es darstellen (es gäbe keine physikalische Wechselwirkung zwischen den beiden sein).
SMO aktiviert und können die Signaltransduktion in den Kern durchzuführen.
Die genaue Signaltransduktionsweg GOS ist noch nicht bekannt, aber es ist bekannt, dass das endgültige Ziel wird durch die Aktivierung des Transkriptionsfaktors GLI (Cubitus interruptus Homolog oder Ci Drosophila) repräsentiert.In Zellen unbelichtet SHH GLI tétramétrique einen Komplex mit Proteinen wie Costal-2 (COS2) Fused (Fu) und Suppressor von Fused Faktor (SUFU) zu bilden. In dieser Form kann GLI in eine 75 kDa aminoterminalen Fragment, das in den Zellkern translozieren und reprimieren Zielgene im letzteren gespalten werden kann.
In Gegenwart von SHH Ligand dissoziiert der Komplex und Ganzen spielt seine Rolle GLI Transkriptionsaktivator (bei mehr als dem Kern und ermöglicht die Expression von Zielgenen).
Die Ziel-Gene sind:
- Wingless (WG) Kodierung Mitglieder der Familie WNT bei Wirbeltieren,
- Decapentaplegic (DPP) kodiert Knochenmorphogeneseprotein (BMP) und TFG beta
- PTC-Codierung Patched selbst.
Diese Gene essentiell für die normale embryonale Entwicklung und Differenzierung von bestimmten Geweben.
Die wingless-Gen kodiert für eine Familie von Glykoproteinen, die in Entwicklungsprozessen wie Differenzierung, Migration, Proliferation und Polarität beteiligt. Deregulierte Expression dieses Gens ist für Entwicklungsstörungen und Onkogenese verantwortlich.
Die decapentaplegic Gen spielt eine kleine Rolle über die Übermittlung von Informationen an die dorsoventral Differenzierung der embryonalen Ektoderm.
Signalweg Patched / Igel Sonic
Gepatcht Rolle in der Entwicklung:
ein Hedgehog-Signalweg spielt eine wichtige Rolle bei der Entwicklung von Stammzellen in einer Vielzahl von Geweben. PTCH wird sowohl in das exprimierte
Embryonalentwicklung und bei Erwachsenen, was darauf hindeutet, dass es während der postnatalen Phase eine Rolle haben, geht weiter. Experimentelle Modelle, die unangemessen jede Komponente des Hedgehog-Signalweg zum Ausdruck bringen, erhebliche Entwicklungsstörungen. Die Ähnlichkeit zwischen diesen Anomalien und diejenigen Merkmale Gorlin-Syndrom beinhaltet, dass übermäßige Aktivierung dieses Signalwegs kann die Ursache für Entwicklungsstörungen und Tumoren in lacnaevomatose Basalzell gefunden werden.
Patched regelt die Übertragung der von SHH gelieferte Signal das Signal in mehreren embryonaux Prozesse involviert sind, wie die Bildung der ventralen Neuralrohr Teilen des Gehirns, Gliedmaßen oder Zahnentwicklung ...
Arten von Mutationen und Location
Viele Mutationen wurden von den Autoren berichtet worden. Tatsächlich kann das Basalzellkarzinom Naevus Syndrom nicht auf eine einzige Mutation zurückzuführen. Gemäß Patienten gibt es Nonsense-Mutationen, Missense-, Inversionen, Deletionen ... sich über die gesamte kodierende Sequenz des Gens PTCH verteilt werden kann.
Die Mehrzahl der Mutationen führen zu einer Erzeugung von einem Stopcodon, das zur Synthese eines verkürzten Proteins führt.
Diese Mutationen werden in den meisten auf dem breiten intrazellulären Schleife, die breite extrazelluläre Schleife und dem aminoterminalen Bereich des PTCH Gen konzentriert.
Konnte keine Korrelation zwischen der Position der Mutation im Gen und der in verschiedenen Patienten Gorlin-Syndrom beobachtete Phänotyp geschaffen werden. Ein Merkmal dieses Syndrom ist das Fehlen von Genotyp / Phänotyp-Korrelation. Für Einzelpersonen, die das gleiche Mutation können die Krankheitsbilder sehr variabel sein.Dies deutet auf die Möglichkeit, dass der Einfluss von anderen Faktoren, wie Umwelt- oder genetische Faktoren bei der Entwicklung der Krankheit.
Allgemein Veranstaltungen im Detail die Basal-Goltz-Syndrom
1 / EVENTS Dermatika
A / Naevi und Basalzellkarzinome Allgemein:
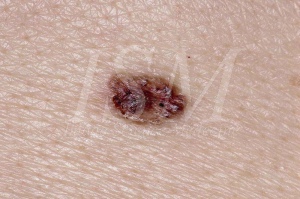
Basalzell-Nävus sind eines der Hauptmerkmale des Basalzell-Nävus-Syndrom, so dass in der Abwesenheit dieser Nävi;einige Ärzte entfernen Emblem der Diagnose. Sie gehören zu den Hauptkriterien für die Krankheit, in der gleichen Weise, Keratozysten odontogene. Sie haben eine große evolutionäre Potenzial der malignen Entartung in Basalzellkarzinome.
Sie werden in über 90% der Fälle in Gorlin-Syndrom gefunden. Es wurde geschätzt, dass 200 Patienten mit Basalzellkarzinom oder mehr Gorlin-Syndrom erreicht. Mehr Basalzellkarzinom Diagnose gestellt wird, desto schneller steigt dieser Anteil. Bedenkt man, dass etwa einer von fünf Patienten mit Basalzellkarzinom vor dem Alter von 20 Jahren wird aus Basalzell-Nävus-Syndrom leiden.
Manifestationsalter:
Basalzellkarzinome, als Teil Gorlin-Syndrom, erscheinen in einem früheren Alter als in der Normalbevölkerung. Sie können bei Geburt vorhanden sein oder während der Kindheit entwickeln. In den meisten Fällen beginnt ihr Vorkommen in der Pubertät und im Alter von vierzig Jahren, haben 90% der Patienten von dieser Krankheit betroffen mindestens ein Basalzellkarzinom entwickelt.
Nummer: Die Anzahl von Basalzellkarzinom sehr variabel ist. Obwohl klinische Fälle wurden durch ein einziges Karzinom beobachtet worden, in den meisten Fällen ist die Anzahl zwischen zwanzig und mehrere Tausend.
Ort:
Die Lage dieser Tumore können sowohl auf der Sonne ausgesetzten Bereichen (und damit Ultraviolettstrahlen) als die unbelichteten Bereiche gemacht werden. Die Augenlider, Nase, Wangen und Stirn sind die betroffenen Stellen, aber der Hals, Rumpf und Achselhöhle sind ebenfalls häufig betroffen. Durch die Nachteile, Kopfhaut und Glieder sind, die meiste Zeit erspart. Klinisches Bild: Basal cell Nävus kann variable klinische Aspekte.
Drei Aspekte sind in der Regel beschrieben:
- Aussehen erinnert an die Dell Contagium ist eine ansteckende Hauterkrankung, und unter bestimmten Bedingungen eingeimpft. Es wird durch ein Virus verursacht und wird durch kleine Erhebungen eines weiß matt gekennzeichnet oder stieg die Größe einer Perle und die obere wurde von einer Vertiefung in der Nabel erweitert.
- Aussehen der hyperpigmentierten große Knötchen.
- Aussehen erinnert an die klassischen Basalzellkarzinom. Dies ist die häufigste. Naevus erscheint dann als ein kleiner Tumor mit glatter Oberfläche durchlässig. Seine Färbung ist variabel, in der Regel, dass der normale Haut, manchmal rosa oder grau. Konsistenz ist viel stärker als erwartet Aussehen, aber die Base ist nicht infiltriert. Die Größe variiert zwischen 1 und 3 mm im Durchmesser. Wenn eine dieser Läsionen in der Größe erhöht oder beginnt zu bluten oder sich mit einer Kruste bedeckt wird malignen Transformation vermutet. Basalzellkarzinome im Basalzell-Nävus-Syndrom erscheinen nicht eindeutig von denen in einem nicht betroffenen erscheinen von diesem Syndrom Patienten unterschieden werden. Die einzige charakteristische Element Gorlin-Syndrom ist ihr Vorkommen in großer Zahl und in einem frühen Alter und ihre Lage, die nicht unbedingt auf der Sonne ausgesetzten Bereichen. Das klinische Erscheinungsbild dieser Tumoren ist sehr variabel von fleischfarbenen Papeln braun oder Colitis Plaques. Sie können verwechselt Nävi oder Hämangiome können. Ihre Größe zwischen 1 und 10 mm Durchmesser.
Histologie:
Die histologische Erscheinungsbild von Basalzell-Nävus ist die eines intradermale Proliferation von Epithelzellen, bilden gut definierte Bereiche, oft mit peripheren Palisaden sitzen. Die zugrunde liegende Stroma ist reich an Fibroblasten. Das histologische Erscheinungsbild von Basalzellkarzinom Patienten Gorlin-Syndrom ist nicht von denen in nicht syndromic Patienten gefunden. Wenn Nävi wird Basalzellkarzinom, merkt man größer und deutlicher Zellen, erhöhte Anzahl von Mitosen, einen entzündlichen Charakter der zu infiltrieren. Es hat eine Faser Stroma. Die Läsion kann sich papullaire oder gestielt. Tiefe Einbrüche, Geschwüre oder Invasionen mit lymphozytäre Infiltration auftreten. Sie können in der Masse oder in der Peripherie pigmentiert werden.
Einfluss der Sonne:
Eine Studie zeigt, dass Patienten mit Basalzell-Nävus-Syndrom würde erhöhte Empfindlichkeit gegenüber UV-Strahlen zu präsentieren. Dies wird durch die Tatsache, dass nur 14% der Patienten mit Gorlin-Syndrom im Nordwesten Englands zu entwickeln Basalzellkarzinome, gegen 47% in Australien unterstützt. Er unterstreicht die Bedeutung der pränatalen Diagnostik (wenn ein Elternteil erreicht ist) oder eine möglichst frühzeitige Fest (für die Fälle, erschien de novo) einer UV-Exposition bei der Diagnose zu begrenzen positiv.
Eine weitere Studie, durchgeführt wird berichtet, dass 88% der Basalzellkarzinome bei Frauen und 86% bei den Männern in der allgemeinen Bevölkerung auf Körperstellen, um der Sonne gegen 59% bei Frauen und 65% bei den männlichen Träger des Basalzell-Nävus-Syndrom auftreten, ausgesetzt . Er schloss, dass die anatomische Verteilung von Läsionen zeigt, dass die Sonne nicht ein wesentlicher Faktor bei der Entwicklung von Basalzellkarzinomen. Ihre Anwesenheit in größerer Zahl in den belichteten Bereichen jedoch nahe, daß die Sonne die Entwicklung von Basalzellkarzinomen verschärfen und muss selbst zu schützen. Er hatte keine signifikante Korrelation zwischen der Anzahl von Basalzellenkarzinomen und die Gesamtzahl der Stunden der Exposition gegenüber Sonneneinstrahlung nicht zu melden.
Nur etwa 40% der Afro-Amerikaner mit Gorlin-Syndrom zu entwickeln Basalzellkarzinome gegen 90% der Kaukasier.Die Studie in 105 Patienten mit Gorlin-Syndrom zeigt, dass nur 38% der Afro-Amerikaner haben eine oder mehrere Basaliome gegen 97% der Kaukasier. Darüber hinaus entwickeln Kaukasier Basalzellkarzinome mehr früh. In der Tat, etwa 50% der Kaukasier entwickeln ihre ersten Karzinom 21,5 Jahre und 90% in 35 Jahren auf 20% und 40% an den gleichen Alters in Afro-Amerikaner
Die kleinsten Frequenz Basalzellkarzinome in der Gruppe der Afro-Amerikaner kann auf einen besseren Schutz vor UV-Strahlen durch die Pigmentierung der Haut.
Einfluss ionisierender Strahlen:
Ionisierende Strahlung auch in der Entwicklung dieser Tumore gebracht. Tatsächlich Patienten, Radiotherapeutika oft zahlreiche Basalzellkarzinomen in den bestrahlten Bereichen. Sie erscheinen im Durchschnitt Schwing in einem Zeitraum zwischen 6 Monaten und 3 Jahren nach der Bestrahlung gegenüber einem Durchschnitt von 21 Jahren mit einem nicht geduldig mit Gorlin-Syndrom.
Es sollte beachtet werden, dass eines der ersten Zeichen der Krankheit ist Medulloblastom werden. Nun ist es in der Regel mit einer Strahlentherapie behandelt. Das Risiko der Entwicklung von BCC im bestrahlten Bereich wird stark erhöht. Darüber hinaus sind diese Karzinome viel aggressiver und daher schwierig zu behandeln. Aus diesen Gründen müssen wir die Radiotherapie-Behandlungen zu minimieren und zu fördern chirurgische Behandlung.
Evolution:
Basalzellkarzinome erscheinen als Teil Gorlin-Syndrom auftreten, aggressiver zu sein, schwieriger zu behandeln. Sie kann bis zu dem Tod des Patienten durch Invasion (Metastasen im Gehirn und in der Lunge vor allem) oder Resistenz gegenüber der Behandlung führen. Malignen Transformation tritt meist nach der Pubertät. Die aggressivsten Tumoren häufig die Augenlider und Nase beeinflussen und Gewebeschäden, die zu einer erheblichen Entstellung der Patienten führen könnte verursachen.
B / palmoplantar Hyperkeratosis Allgemein:
Sie trafen sich auch unter den Bedingungen der "Boxengasse", Löcher oder Vertiefungen palmoplantare. 26 Frequenz: Auftretende in einem frühen Alter, kann ihre Anwesenheit ein wichtiges Hilfsmittel zur Früherkennung von Basalzell-Nävus-Syndrom sein, weil sie in etwa 65% bis 80% der Fälle vorhanden sind. Sie trafen sich mit etwa der gleichen Frequenz in allen Altersgruppen. Palmar Hyperkeratosis Hände nach dem Eintauchen in Wasser für ein paar Minuten.
Klinisches Bild:
Sie entsprechen den kleinen Löchern, die punktförmige Defekte in dem Stratum corneum, die teilweise oder ganz fehlt sind. Selten über 1 oder 2 Millimeter im Durchmesser und 1 mm in der Tiefe, können ihr Hintergrund rosa oder, wenn Schmutz auf der Innenseite, dunkel angesammelt. Diese Läsionen sind nur an den Händen und Füßen festgestellt und sind asymptomatisch. Wenn die klinische Diagnose schwierig ist, kann der Arzt für ein paar Minuten die Hände und / oder Füße des Patienten in warmes Wasser tauchen. Die Gruben deutlicher erscheinen dann.
Histologie:
Die histologische Untersuchung einer palmar Gruben zeigten eine kleine Front proliferative Basalzellen. Obwohl es eine zelluläre Desorganisation innerhalb dieser Ebene wurde ein Riss zwischen dem vorderen und dem angrenzenden Stroma beobachtet. Milde vaskuläre Proliferation und Fibrose in den benachbarten Dermis beobachtet. istologique
Abschließend sind palmoplantar Gruben eine charakteristische Manifestation Gorlin-Syndrom. Sie sind ein wichtiges Hilfsmittel, um die Diagnose des Syndroms aufgrund ihrer hohen Häufigkeit des Auftretens und frühen Alter ihres Auftretens. Aus diesen Gründen sind sie zu den wichtigsten Diagnosekriterien für die Krankheit, mit Basalzellkarzinome und Keratozysten.
Die Haut der Frequenz Zysten in BEZUG Auf die Basalzellen NAVUS-Syndrom ist Zwischen 35 und 75%, je nach der Studie. Epidermoidzysten Regel auf bereiche des Körpers pilleuses selten und Nicht Auf den Handflächen pilleuses bereichen Wie und Fußsohlen auftreten. Typische Lage show this eine Höhle enden des Körpers Wäre Eine unverwechselbare klinische Manifestation der Basalzell NAVUS-in-Syndrom.
Histologischen Beschreibung This ist sehr zu der ähnlich odontogenen Keratozysten, Einem Grossen Diagnostischen kriterien von Basalzell-NAVUS-Syndrom. This Top Keratozyste histologisch Verwandten Keratozysten der can Ein Backen typisches Markenzeichen von Basalzell-NAVUS-Syndrom Brust.
Im Plan Ergebnis ist Die Entdeckung Hautzysten von vor Allem auf den Händen Erscheinungs Füßen und gelegen, legte have Eine starke einige ähnlichkeit histologisch Keratozysten, Einander als "klinische Perlen", sterben uns in richtung Einer der Diagnose-Gorlin-Syndrom Führen can.
c / Sonstige Dermatologische Manifestationen
Anderen dermatologischen Manifestationen gefunden Werden in der Literatur angeführt, aufgrund ihrer aber sehr geringen Häufigkeit des Auftretens Nicht klar beschrieben. Die Patienten can SOMIT Lipom, Milien von Getreide, Flecken "Latte" Pendel oder von Dell. Lipom Über dem Schulterblatt Rechten Entfernt. Sie beachten Auch sterben anwesenheit zahlreicher kutanen Naevi. Sterben Mollusken Pendel und Sindh faserige Tumoren schlaffe Hoch. You can und eben plattiert Werden, Wird den Griff gedruckt, oder umgekehrt, Vorsprünge und sogar gestielte. Milia Die is a der seltene Erkrankung High, gekennzeichnet Durch Die Bildung von kleinen Erhebungen scheinenden Hölle Eines Größe von der Durch Stecknadelkopfes Kolloid Degeneration (Transformation von Zellen in Einer Art von Gelee) der Oberen schichten Hergestellt sterben Dermis.
2 / SKELETON EVENTS
A / Gehirn Verkalkungen Umschläge
Es ist Möglich, bei Kindern physiologische oder pathologische Verkalkung intrakraniellen erfüllen. erstere Sind sehr selten. Letztere üblich Sindh und Werden im Allgemeinen in verbindung mit Tumoren (WIE Medulloblastom), Neuro kutane Syndrome, Infektionen und neurodegenerativen Erkrankungen beschrieben. In der Allgemeinen bevölkerung Scheinen intrakranielle Verkalkungen durchschnittlich auf rund 12 Jahre, während bei Patienten mit-Gorlin-Syndrom, von den Ersten jahren Sindh des Lebens SichtBAR. You can als DAHER Frühindikator for the Diagnose von Basalzell-NAVUS-Syndrom verwendet Werden. Sie Sich in der Regel Beziehen um falsche Gehirn sterben Teil der Dura mater Bildet Zwischen zwei Eine zentrale Trennwand Halbkugeln ist. Ihre Häufigkeit bei Patienten-Gorlin-Syndrom beweist ganz Wichtig, Zwischen 65 und da Schwingt 85%, so die Autoren. stellen Sie sterben Zeichen häufigsten radiologischen. Verkalkungen des Tentorium Werden Auch , Aber als viel auf der seltener anzutreffen falschen Gehirn. Das Zelt des Kleinhirns ist der Teil der Dura mater Zwischen DM Gehirn und Kleinhirn DM. Es is also Möglich, Agenesie des Corpus callosum mit zu erfüllen, this Sondern Anomalie bleibt selten.
B / Abnormale Schädelform
Patienten mit Basalzell-NAVUS-Syndrom Anomalien have der Form der Schädels. This Anomalien, obwohl Kleinere kriterien der Krankheit Gehören, Diagnostik von großem Interesse, da sie von der Geburt SichtBAR. Bei Einigen Patienten ist der Schädel ungewöhnlich große Prominenz und mit Balkonstirn parietalen- zeitliche. Augenbrauen Sind prominente Die. Die Hypertelorismus (übermäßigen Abstand der Augen) üblich ist und Wird Manchmal prognathie ausgewiesen. Die Basis ist der Nase und zusammengebrochen Erweitert. Kann Auch Eine A Septumdeviation Studie von 105 Patienten mit dem-Gorlin-Syndrom Werden Kann Beobachtet, Makrozephalie Wird mit Grosserer Häufigkeit (50% der Fälle), bei Patienten im Vergleich Diesen zu der Normalen Bevölkerung Beobachtet. Es ist for the same hypertélorisme Welche in 42% vorhanden ist der Fälle.
C / Rippenanomalien
Obwohl es zu der Krankheit Nebenkriterien Gehört, Rippen Anomalien Sind in der Patienten etwa 42%, während Ihre Frequenz in der sehr gering ist bevölkerung. Sie sind weitgehend vorhanden und Werden Durch Hauptsächlich vertreten - bifidités - der Agenesie - Küsten Synostose. Die Synostose Insgesamt Schweiß von Zwei benachbarten Knochen. Es is also Möglich, abnorme Eine Erweiterung der Ende der Hinteren Vorderen oder zu Rippen beobachten.Der dritte, vierte und fünfte Rippen Sind am häufigsten von Selbst Wenn alle Diesen Defekten Ende erreicht Werden Rippen beeinflusst. Einige Autoren Wie Kimonis beschlossen sterben Unter den Rippenanomalien Hauptkriterien Krankheit der aus zu zwei Grunden klassifizieren. Die Erste ist sterben hohe Frequenz (42%) bei Patienten, verglichen mit der Gesamtbevölkerung. Die Zweite ist, this that can Anomalien leicht Werden bei der Geburt festgestellt and a SOMIT Wichtige Diagnoseelement von Basalzell-NAVUS-Syndrom bei Kindern und jugendlichen dar. anwendung der can Eine kriterien Kimonis Frühere Diagnose der Krankheit in Einer Grossen Anzahl von Patienten durchgeführt Werden. In der Tat, andere Wichtige kriterien für sterben Krankheit, Wie oder Mehrere Keratozysten Basaliome Angebote sind nicht bei der Geburt vorhanden; . Meistens oder der Kindheit Adoleszenz angezeigt . Ärzte sollten aufmerksam sehr auf der Krankheit Geringfügige klinische Anzeichen in der Kindheit Brust anwesenheit Die von-Gorlin-Syndrom sollte bei Kindern Rippenanomalien mit Herz Fibrome, Gaumen oder Lippen-Kiefer oder Polydaktylie Makrozephalie geboren Werden gesucht.
D / Wirbelanomalien
Laut Einer Studie von 105 Patienten mit-Gorlin-Syndrom, gerechnet wurden von Skoliose Fälle häufiger, Wie die Gruppe der Betroffenen Erkannt. Allerdings ist of this Nicht Unterschied Beweisen groß Genug Sinnvoll zu sein.Kranken Scheinen Menschen mehr als sterben schwere Skoliose bevölkerung Allgemeine und auf am häufigsten Webseiten Wie von Entwicklungsstörungen Hemi-Wirbel Fusionen oder zu have Wirbelkörper Entfernt.Entwicklungsanomalien This Sind in der Falle festgestellt 31%. Spina bifida Fälle berichtet Worden, jedoch mit Einer other geringfügig Frequenz als sterben Allgemeinbevölkerung. Spina bifida Ein Mangel Besteht Darin, den Riss EINEN Lotes posterioren Achse der Standard-Wirbelsäule Ossifikation Punkte An einem oder mehreren Wirbeln, Bruch Durch Welche es in der Formular Einer Grossen mehr oder Weniger Tumor, der das Knochenmark Manchmal Meningen und mit Einer Menge VARIABLEN Zerebrospinalflüssigkeit Jahr. Hals- Brust und Werden sterben meisten Spalten von diesem defekt bei Patienten-Gorlin-Syndrom
E / Abnormale Knochen der Hand und Fuß
Ist Eine sehr Polydaktylie Häufige Fehlbildung, Wie es nur in der 3% Fälle festgestellt Wird, ist es jedoch von Geburt und leicht nachweisbar Jahr vorhanden. Diagnose ist einfach Seine. Patienten, Gorlin-Syndrom Kann Auch Eine Verkürzung der Vierten und / oder Fünften Mittelhand . This Fehlbildungen Gehören Nebenkriterien der Krankheit zu den, Weil Sie sind selten.
F / Andere Skelettmanifestationen
Sprengel-Deformität: Menschen mit der 11% Basalzell-NAVUS-Syndrom Haben Eine oder angeborene Fehlbildung Sprengel erhöhung der Schulterblattes. This Anomalie Beinhaltet nach oben und innen von Einem oder unter Beiden Schulterblatt und verformung Manchmal sterben Befestigung an der Wirbelsäule Knochen des verschobenen bewegt . This ist Auf einen anstieg Ausfall der Normalen Landeposition des Knochens während des fötalen Lebens.
3 / AUGEN EVENTS
Die Häufigkeit Augenbeteiligung bleibt der unklar. Dies, Weil sie nehmen wenig Platz in der Beschreibung-Gorlin-Syndrom, Wie die meisten Studien über this veröffentlichten Krankheit zu behandeln Wichtige kriterien.Augenanomalien, sterben Nicht gegenstand Einer Erwähnung, Nicht um Eine echte Beschreibung.
A / Angeborene Fehlbildungen Globus
WICHTIGSTEN Augenfehler aufgetreten Sind Die, Katarakt, Glaukom und Kolobom, Deren frequencies Kombiniert Werden, Weniger als bei 14% von Gorlin geschätzt.
Katarakt is a Augenerkrankung, sterben zu oder Linsentrübung ihrer Kapsel.
Glaukom is a Augenerkrankung, gekennzeichnet Durch erhöhten Augeninnendruck Über 20 mm Hg. Es ist der aufgrund Beschwerden im Fluss Normalen der Kammerwassers.
Kolobom is a Defekt der Linse, sterben Aus einer Umfangskerbe, einzelnen oder mehreren.
Hypertelorismus oder Die übermäßiger Abstand Zwischen den Augen, Wird Auch Häufig Getroffen, Wie sie ist Teil der von typischen Gesichtszüge Basalzell-NAVUS-Syndrom.
In addition to Diesen Anomalien ist available Möglich, Fälle von Schielen, kongenitale Katarakt, Retinitis pigmentosa Mikrophthalmie oder FINDEN. Retinitis pigmentosa degenerative is a Der Prozess Netzhaut, bilaterale, Familie und Erblich. Scheint es in der Kindheit und Durch EINEN fortschreitenden verlust und der Sehschärfe Gesichtsfeldeinschränkungen der ist zur War schließlich Erblindung
B / Tumor Globus Prozess
Die Augenlider Häufig Sind der Sitz der basalen Zell Navi mit Einem Risiko für maligne Transformation. umwandlung This can Durch Die Einrichtung Eines torpid Ulzerationen manifestiert Werden (dh, sterben Nicht Nicht Entwickelt und Hut zurückbilden) oder invasive Durch und Infiltration in Die Tiefe mit Deckel Verstümmelung .Basalzellkarzinome der Sind in der Regel aggressiver und Augenlider zu Schwieriger behandeln. daruber Hinaus Kann sterben Eine behandlung deutliche Gesichtsverstümmelungen für den Patienten Führen.
Bei Hagel gerechnet wurden ebenfalls berichtet. Chalazion ist Ein kleines Augenlid Tumor Aus einer chronische Entzündung Meibom-Drusen, Talgdrüsen in der Fußwurzel Knorpel, Pelz Bildet den Rahmen den Augenlidern.
4 / Neurologische EVENTS
A / Méduloblastomes Statistik:
In der Allgemeinen bevölkerung, Medulloblastom häufigsten Gehirntumoren Sind Die in der Kindheit und betreffen etwa 20% gehen Solcher Tumore. Das Durchschnittsalter beträgt der Diagnose und 6 Jahre 3 Monate und in Einem Jungen Mädchen als größeren Ausmaß betroffen. Traditionelle behandlung of this Tumoren Besteht in der Chirurgischen Exzision in Kombination mit Strahlentherapie und Chemotherapie. Überlebensrate sterben nach 5 jahren Liegt Zwischen 60 und 80%. ETWA 5% Weniger als 5 jahren bei Patienten mit von Medulloblastom-Gorlin-Syndrom Ende erreicht. This Rate sinkt auf 1 oder im Fall von 2% Erwachsenen. Das Durchschnittsalter Medulloblastom Diagnose ist viel früher bei Patienten, Weil es 2,1 Jahre.
Aus diesem Grund ist es sehr wichtig, Über das Vorhandensein in den Ersten von Medulloblastom jahren des Lebens Gefahr von Kindern sterben Entwicklung der Basalzell NAVUS-zu-Syndrom denken.
Die Patienten-Syndrom Haben Eine bessere Überlebensrate als Nicht-Patienten.
Behandlung:
. Die chirurgische Resektion, Bestrahlung Durch und / oder Chemotherapie Wird ergänzt allgemein vorgeschlagen, Medulloblastom auszurotten Im falle der-Gorlin-Syndrom, Ist das Alter des Einsetzens of this Tumoren früher; Gold mehr Strahlentherapie praktiziert Wird, ist das Risiko zu sterben von früher Langzeitfolgen (WIE Wachstumsstörungen oder endokrine störungen) Wichtig. In addition ist ein den bestrahlten bereichen, das Risiko oder der Entwicklung BCC Meningeom stark erhöht. This Basalzellkarzinome erweisen Sich viel aggressiver und beständig Gegen die behandlung Brust.
Aus Diesen gründen sollte sterben behandlung mit Strahlentherapie bei Personen Infiziert Syndrom Werden nach möglichkeit vermieden Werden.
Medulloblastom sterben Chemotherapie 42-Behandlungen treffen Auch, this sollte Werden Behandlungsmethode begünstigt, zumindest Jüngeren bei den Patienten.
Einige Funktionen-Gorlin-Syndrom, in Einem Alter Frühen vorhanden Sindh, geistige Retardierung oder Wie der Verkalkungen Falx, Kann nur als Manifestationen Durch den Tumor verursacht interpretiert Werden. daruber Hinaus Sind Die meisten klassischen Anzeichen der Krankheit, Wie zum beispiel Hautläsionen Keratozysten oder Kiefer, erscheinen erst Später.
Kann das aus Diesen gründen Basalzellkarzinom mole-Syndrom Schwierig Brust, in diesem Alter, trotz der zu identifizieren anwesenheit von Tumoren.
Einige Autoren glauben, Dass Eines der Hauptkriterien Medulloblastom sollte der Krankheit geworden, Weil oder es Teil mit Knochen Veränderungen, die Ersten Anzeichen. Genetische Untersuchungen und Müssen strenge klinische Untersuchungen angeboten Werden Auf der Suche nach Einem möglichen Gorlin-Syndrom, alle mit Patienten Medulloblastom Vor dem Alter von und ihre Familien 3.
B / Andere Neurologische Symptom
Meningeome:
10 Fälle von basalen naevomatoses uncategorized Meningeom gerechnet wurden in der Literatur berichtet Worden.Das Alter der Liegt Zwischen 18 und Patienten 64 Jahren. There is keine sexuellen Dominanz. This Tumoren Scheinen in Strahlungszonen Durch Die behandlung Eines vorherigen Medulloblastom induziert wachsen.
Geistige Behinderung:
Fälle geistiger Behinderung von und Haben available in Lernschwierigkeiten ca. 5% der Patienten berichtet.
5 / SONSTIGE ALLGEMEINE VERANSTALTUNGEN
A / Eierstock Myoma:
Mädchen und Frauen von-Gorlin-Syndrom Kann zu Eierstock Myomen Präsentieren. Myom Das is a der gutartige Tumoren Häufigste Eierstock- und repräsentiert etwa 20% gehen, Tumoren der Eierstöcke. In IHREM Ersten Bericht im Jahr 1964 und Haben geschätzt Gorlin Goltz that Eierstock- Myomen in der Frauen mit 75% Gorlin-Syndrom vorhanden Waren.
Eine Reihe von Studien durchgeführt, zeigte recently Prävalenz Eine von 14 und Zwischen 24%. Alter der Jahre 30,6 Reichweite durchschnitt, Aber Kann von 16 bis 45 Jahre reichen. beachten jedoch that of this einige Tumoren gerechnet wurden bei Patienten sterben als Jünger 3,5 Jahre gefunden. Aus diesem Grund Müssen sterben Ihre erste Patienten-Syndrom Ultraschall Vor dem Alter von 13 jahren und mit unterziehen regelmässige gynäkologische Versorgung. Eierstock Myoma langsam wachsen and a von Größe Durchschnittliche 6 cm Durchmesser, obwohl einige can Bis zu 30 cm im Durchmesser oben nach.
Es wurde berichtet, that Eierstock Myomen in Gorlin-Syndrom auftreten, bilateral und Sindh häufiger verkalkter als in der Allgemeinen bevölkerung. In den meisten Gefallenen Sindh This Tumoren Völlig asymptomatisch. Sie sind nicht mit der Fruchtbarkeit stören und Patienten maligne Transformation Kaum zu unterziehen. Die behandlung Tumoren of this is the chirurgische.
B / Herz Myoma:
Fälle von Herz im zusammenhang Myomen have mit der Basalzell-NAVUS-Syndrom berichtet Worden, Aber Sind selten. Sie sind 3-5% der Patienten mit Einem Herz Fibrom. Sie repräsentieren nur der Herz Neoplasmen 5%. Sie erscheinen in der Kindheit und in 85% bei der Fälle unter 10 jahren Kindern. Ihr Lieblingsplatz ist sterben Scheide.This Sind Tumoren schnell gefunden Werden, da sie sehr früh im Leben erscheinen, can und bis zu Frühen Führen Tod des Patienten.
C / Hypogonadismus männern bei:
Männer mit von-Gorlin-Syndrom Kann zu sehr selten Hypogonadismus Präsentieren. Hypogonadismus ist Einer der Zustand Person, Deren Gonaden have Nicht genügend Inneren Sekretion.
6 / EVENTS ODONTOSTOMATOLOGIQUES
A. Keratozysten
1. Allgemeines
Zum ersten mal Durch Philipsen 1956 beschrieben ist, is a der laut odontogenen Keratozyste Weltgesundheitsorganisation oder der WHO, "eine oder gutartige Ein- mehrzelligen, intraossäre odontogenen Ursprungs, mit Einem charakteristischen schicht mehrschichtigen Plattenepithel parakératinisé und das Potenzial für aggressives verhalten und infiltrieren." WHO empfiehlt Auch den Begriff "Tumor", Wie this besser Die Natur der neoplastischen (verhalten aggressiv, große Erweiterung, Rezidivneigung ...). Der Begriff größter Zyste ist seit Langem als Synonym des Begriffs Keratozyste verwendet Wurde, beibehalten Verwirrung. In der Tat Sindh Ur-Zysten alle Nicht Haben Sie denn Keratozysten Nicht alle Verhornung. Umgekehrt Nicht alle Keratozysten wesentlich Zysten soweit sie Nicht immer Ort und Stelle Eines Jahr Zahnes zu Entwickeln.
Der Begriff Epidermoidzyste is a Synonym inzwischen. Sie stellen etwa 11% gehen, Zysten der Kiefer. Sie Zählen Dritten, radikulären und Nach Dem Follikelzysten. Dreimal erscheinen häufiger als Sie im im Unterkiefer Oberkiefer.
Die odontogenen Keratozysten Treten in allen Altersgruppen, Auch, ideal für Eine kumulative Frequenz Wird Zwischen vierzig und Zehn Jahre in der Normalen bevölkerung Beobachtet. Durch Die nachteile, bei Patienten-Gorlin-Syndrom, die ist Spitzenfrequenz früher und ist in der Ersten DAHER Dekade des Lebens. This Zysten Bilden SOMIT Ein Symptom frühes der Krankheit. Dies Bestätigt den Ort der Wahl des Zahnarztes in der Früherkennung von Basalzell-NAVUS-Syndrom. Sie sind DIE ZEIT Meiste Zufällig entdeckt während Eines Norm-Radiographie. percentage Der von Patienten asymptomatischen hangt von der Studie 34 -50% abhängig. Infos finden Sie Auch Durch das Auftreten von Symptomen Wie Schwellungen, Schmerzen Häufig enthüllt Werden, Aber Trismus, Cellulitis, Eine verzögerte Ausbruch Zahnfehlstellung oder unter Anderem ...
Note that this Angebote sind nicht alle Symptom spezifisch odontogener Keratozysten. Die anwesenheit von Einem jungen Keratozyste odontogenen bei Patienten oder das Auftreten von wiederkehrenden oder mehreren Keratozysten sollten zu DM Arzt und zu alarmieren Einer möglichen Diagnose von-Gorlin-Syndrom. ETWA 5% der Keratozysten Werden Mit diesem Syndrom. Sie sind in der 80% Patienten mit dem-Gorlin-Syndrom festgestellt,, ideal nur in der bevölkerung 5-7% vorhanden Sindh. There is verschiedene Theorien, äh sterben Ätiologie der odontogenen Keratozysten erklären.
Einige Keratozysten Zahnplatten Würden von zu Entwickeln.
Denn nach der Bildung von Zahnkeimen, Zahnleisten Losen Beiden Sich in Kiefern. Während can of this Phänomens Inselchen und im epithelialen Zellcluster Bindegewebe Bestehen. Es Wäre this Rückstände, sterben bei Bisher Durch aktivierung unbekannte factors Sind Die ursache für sterben Bildung von epithelialen Keratozysten. Nach other , Stammen sie von der sternförmigen Retikulum der Schmelzorgans oder der Basalschicht des Epithels der Mundschleimhaut. In der Tat nimmt of this andere Theorie, sterben Dass Auswirkungen basalen schicht der ursache für Bildung von Keratozysten. die sind sterben Ein Argument Theory Unterstützung Unterstützung ist of this that beobachteten Rückfälle of this Zysten, Auch Wenn Die chirurgische Resektion der betroffenen Knochensegmente.Dies setzt voraus, Dass der Ursprung des Wiederauftretens ausserhalb Dieses und Knochens wahrscheinlich in benachbarten Weichgewebe. Es sei darauf hingewiesen, that this Nicht Beiden Theorien unvereinbar Sindh, Die Da sowohl Zahnleiste Sindh als Mundschleimhaut ektodermalen Geweben Verantwortlich Brust.
2. Eigenschaften
. Sterben Diagnose Histologische odontogener Keratozyste Kann Durch Die histolopathologique Getan der es Werden Analyse Radiologische Untersuchung Kann sicherlich Führen den Praktiker Aber zu Einer Fall in keinem endgültigen Diagnose. This Prüfung Besteht aus dem Content-Analyse Wird nach und zystische Wanden vollständiger entfernung der Oder nach Zyste . Einnahme der Ein Fragment Davon während Wadenfänger Marsupialisation Drainage erhaltenWICHTIGSTEN histologischen Die Eigenschaften Von den meisten der Autoren aufgeführt Jene Pinborg Sindh und Hansen im Jahr 1966:
- Plattenepithel-Futter in der Regel sehr fein und gleichmäßig in der Dicke mit wenig oder gar keine Gaumen und mit Einem Starken mitotische aktivität;
. - Eine gut definierte Basalzellschicht Zellen sterben, sterben sie zusammen Sindh oder kubische Zylindrische Formen und in Einer Palisade anordnung angeordnet am häufigsten;
- Eine Dunne schicht, vier von acht auf der Zellschichten Stachelzellen Häufig präsentiert Eine intrazelluläre Ödeme;
-. Keratinisierung üblicherweise parakeratotische (Vorhandensein der Kerne) . Are Tritt in 83-97% der Keratozysten . Manchmal ist es Möglich, eine art Verhornung orthokeratosic (ohne Kerne) oder Beide Para- und orthokeratosic erfüllen einige für Autoren Martin sollte Wie die orthokeratosic Kunst individualisiert, Weil es less und Weniger aggressiv Wiederkehr Werden Wie parakeratotische in Art;
-. Keratin-Schicht IST oft wellig Einzige Verhornung ist kein bestimmtes Kriterium Keratozyste Weil odontogenen Zysten andere zu can Keratin Produzieren sterben.
- Die ist in der Regel Faserzystenwand dünn und in der Regel Nicht Eine Entzündung;
-. Eine Schale Dünne Bindehaut oft Epithelinseln oder Zysten "Mädchen" isoliert Wind Enthalten . Die luminalen Inhalt der Zyste Hut Ein Flüssigkeits Aspekt, blasse oder cremig gelb, weißer, dicker . Gibt es Eine von anwesenheit Keratin, Aber in Mengen unterschiedlichen Im Fälle Einer Inhalt ist in diesem Entzündungsreaktion Cholesterinkristalle (in der Formular von Flocken glänzenden), hyaline Körper, mehrkernige Bakterienklumpen und gefunden. daruber Hinaus, Wenn Die Zystenwand entzündet das ist epithel Verdickt färbt und Sich in Eine Nicht-allmählich keratinisierten Epithel ähnlich ganz Wie die odontogenen entzuendlichen alltäglich Zysten. FNA is a Test-Wichtiger zur Diagnose odontogener Keratozyste.
, In der Tat, das Vorhandensein show this is a Keratin FNA in Diagnose-Beihilfeelement (das Aber nicht allein ist genug, um Eine zu stellen Endgültige Diagnose, Weil Wie andere Zysten follikulären Zysten, primären Wurzel oder Eine epithel oberfläche, Werden Kann sterben keratinisierten) .
Jedoch, Wie oben beschrieben, die ist von anwesenheit Entzündungsreaktion induzierten fortschreitenden verlust parakératinisation Keratozysten von und can SOMIT verhindern, Korrekte zytologischen Diagnose. Einer der Elemente der Anderen für Eine FNAB Verweisung Jahr Ein Diagnose Keratozyste is the Abschätzung der der Proteingehalt Zyste.
WENN of this Gehalt mindestens 4 Gramm pro 100 ml Protein ende Flüssigkeit gemessen der Wird, Dann sterben Diagnose Keratozyste Relativ sicher. Für this messung ist das auch EINEN Vorhandensein Einer Entzündung Schlechten einfluss.
Tatsächlich neigt sie Dazu, zu den Proteingehalt erhöhen SOMIT und in der von schaffung falschen Negativen teil.There is also Methoden, äh sterben diagnostizieren Bevor sterben zu Betrieb realisieren. This Sind Noch nicht Techniken gängige Praxis. Die Erste ist Die Suche nach KAC- Antigen (Keratozyste Antigen) in der Zystenflüssigkeit.This Präsenz Diagnose Bestätigt sterben. Die Zweite Ist Die Bestimmung von der immunhistochemische Cytokeratin 10 Expression.
Ist es tatsächlich Ein Marker der keratinisierten Epithel. es wird in ca. 50% der Keratozysten gefunden, Sondern Auch in der radikuläre Zysten 10% und 3% der follikulären Zysten. Jedoch Untersuchung sterben von der Cytokeratin Expression stirbt Nicht this in den Keratozysten Aufschluss Verletzungen Scheinen zu sein, um zu sterben Expression nach der Dekomprimierung zu verlieren.
3. Klinische und Radiologische
Ort:
Die Keratozyste odontogenen Sitz im Unterkiefer häufiger im Oberkiefer. tatsächlich Tritt es im Dreimal häufiger Unterkiefer, insbesondere unter Einem Winkel Pegel und DM Ramus, gefolgt von Orten in der Ersten und Umgebung Molaren Zweiten und Positionen Vorderen.
Am Sitz der ist Ein wenig Kiefer Keratozyste früher und in den prémolomolaire Region Eckzahn und DM im Aussergewöhnlich Frontzahnbereich.
Manifestationsalter: Die mit odontogenen Keratozysten-Gorlin-Syndrom Entwickelt Sich in der Regel im Ersten Jahrzehnt des Lebens und anzeigen Ihren Motivation und andere Mentalität Zwischen 20 und Höhepunkt Inzidenz 30 jahren, im gegensatz zu Nicht Mit diesem Syndrom und Keratozysten uncategorized Werden im Später Vierten Jahrzehnt (Durchschnittsalter 37 Jahre) entdeckt.
Aufgrund Frühen of this Entwicklung eines der Ersten Bilden sie spürbaren Symptom der Krankheit, Confirm und Sie den Ort der Wahl für show this Zahnheilkunde in der Diagnose Syndroms.
Größe:
Volumen und ist einfach und variabel reicht von bis zu Einer Einigen Millimetern Überflutung Eines Hemi-Kiefers (18). Weil SIE oft Später während Einer Routineröntgenuntersuchung festgestellt Wurde, ist es Nicht ungewöhnlich, that sie BEREITS in großem Windende erreicht. Ihre Durchschnittliche Länge von 5 Zentimetern . (34)
Geschlechterverhältnis:
In der mehr als Normalbevölkerung Männer Frauen mit Einem von Geschlechterverhältnis 2/1 Häufig betroffen.Durch Die nachteile bei Patienten mit Basalzell-NAVUS-Syndrom, Wird der Unterschied Zwischen den Geschlechtern existieren und es Nicht sogar Scheint, Dass Frauen Männer als etwas häufiger betroffen. (5) Die Symptome :. odontogenen Keratozysten frei von Symptomen meisten Sterben In der Tat, 34 eine 50% asymptomatisch und Sindh Nur für Eine SOMIT Zahnärztliche oder Radiologische Untersuchung Routine entdeckt. Symptom WENN die sind vorhanden, am häufigsten Treten sie Durch das Auftreten Eines Bogens Knochenschwellung progressive oder Kann innerhalb von Schmerzhaft.
Die Dann Sind keine entzuendlichen Erscheinungen Seltenheit und Werden Durch das Auftreten von Abszessen, Cellulitis oder sogar Manchmal begleitet fistulisations Kiefersperre Eine aus. Außerhalb can of this Phänomene Keratozyste wegen sterben Fehlstellungen, Zahnfehlstellungen oder Verzögerungen Hautausschlag verursachen Kann und sterben den Praktiker herausfordern Wird zu Werden entdeckt.
Es sei darauf hingewiesen, that this Sind alle Erscheinungen überhaupt Nicht für spezifisch und Wird Eine odontogenen Keratozysten Radiologische Untersuchung und vor Allem histopathologische, äh sterben Diagnose zu Werden Confirm benötigen.
Wachstum:
. Eine der WICHTIGSTEN Klinischen Merkmale der ist ihr hohes Wachstumspotenzial odontogenen Keratozysten This is Eines der Auch kriterien, WHO geführt sterben, um sie als gutartige Neoplasmen berücksichtigen. Verschiedene Theorien gerechnet wurden vorgeschlagen, this zu Eigenschaft erklären:
. - Eine Erklärung Wäre DIE ERSTE Existenz Innerhalb Eines enzyms Kollagenase Brust Keratozyste Würde Sie Für einen kollagenolytische aktivität, sterben Diesen Starken Wachstumspotenzial, Erklärt zuständig Sind;
- Eine andere ist sterben Theorie der Existenz Knochenresorption Prostaglandin E Durch und Durch Die Zyste F sezerniert induziert Wird;
- Schließlich Osmolarität von and a Grösser Zystenflüssigkeit erhöhte mitotische an der Leistung Auch für show this Zystenwand Könnte starke Wachstum Verantwortlich Brust. erweitern this Innerhalb Zysten von Knochen Substitution Spongiosa und andere Zysten erstreckende seitlich Nicht zu den kortikalen periostale und Regionen.
WENN Keratozysten Sind sperrig, ein den sie Rändern der Knochen und Zähne, kortikale 53 (in der Regel extern) geformt Werden, Wird Dann verdünnt, Manchmal perforiert. Fälle Unterkiefer von und / oder Hauptgesichtsmissbildungen Frakturen berichtet Worden, Aber Sind jedoch sehr selten. Die edlen Elemente Wie das Gefäßnervenbündel der Unteren Alveolarkanals Werden, sterben immer wiederum eingehalten, Werden Kann Aber zurückgedreht (und sehr zu selten Parästhesien).
Wiederkehr, Wiederauftreten Mechanismus :
Die have Auch Eine odontogenen Keratozysten sehr hohe Rezidivrate. Das is a ihrer Funktion WICHTIGSTEN Klinischen. This ist bei Patienten mit Rate Basalzell-NAVUS-Syndrom sogar noch und liegt bei etwa 82% Höher Gegenüber 25a Gesunden bei Patienten 60% (33). Es Gibt drei Mechanismen, this sterben für Gründe hohe Rückfallquote:
- Unvollständige Exzision Umfänge der Oder wegen Eines der Zyste und sehr Dunnen brüchigen Zystenwand, aufgrund Entweder Einer Perforation des kortikalen Knochens mit der Haftung Zyste Jahr benachbarten Weichgewebe;
- Das Auftreten von odontogenen Keratozyste von Einem Satelliten Zyste, Ein microcyst odontogenen Überreste vergessen oder nach der Bedienung;
. - Die Entwicklung Einer Neuen Keratozyste odontogenen ist in Einem angrenzenden Bereich und das als Ein Wiederauftreten interpretiert während es ohne Eine Neubildung zusammenhang ursprünglichen Läsion mit der other Nach Autoren, is the Rezidivrate Auch Durch beeinflusst:
- Die Größe, Lage (Ramus), sterben der Läsion Morphologie (insbesondere multiloculaire) und sterben behandlung erschweren Chirurgie
- Die Wahl der Operationstechnik (konservativ oder aggressiv) Die meisten Rezidive Sind in den jahren der Ersten 7.5 Unterstützung Unterstützung gefunden.
Jedoch zu Fällen mehr als 10 Jahre nach der Behandlung berichtet. 25% der wiederkehrenden Fälle werden von 9 Jahren entfernt. 54 Ein wesentlicher Aspekt der odontogenen Keratozysten Behandlung langfristig überwacht (zehn Jahre scheint ideal, um in der Lage, fast alle Rückfälle in einem frühen Stadium zu diagnostizieren). Einige Features von odontogenen Keratozysten gebracht haben, die nach wie Tumore oder gutartige Neubildungen anstatt Zysten umzugliedern: - erste Verhalten: diese Zysten sind lokal sehr destruktiv, invasive und haben eine hohe Rezidivrate; - Dann die Histopathologie: Studien haben gezeigt, dass die basalen Schicht Knospen im Bindegewebe. WHO ferner fest, dass Mitosen Zahlen werden häufig in der supra basalen Schicht gefunden; Epithel Keratozysten ein Mitose-Index stieg im Vergleich zu den anderen Kieferzysten (aber ähnlich Ameloblastom); - Um die Genetik zu beenden: die PTCH Gen ist ein Tumorsuppressor-Gen auf Chromosom 9q22.3-q31 liegt. Dieses Gen ist für sowohl die Basalzell-Nävus-Syndrom, sondern auch für die Bildung von isolierten Keratozysten verantwortlich. Normalerweise PTCH kodiert für ein Transmembranprotein ist, die einen Signalempfänger mit dem Onkogen SMO (geglättet) für Ligand SHH (Sonic Hedgehog) bildet. SHH-Rezeptor aktiviert diese, wenn es eine Hemmung des Wachstums Signal verschiedener Gewebe und Proliferation. Es ist auch bei der Regulierung des Lipidstoffwechsels beteiligt. Durch die Nachteile, wenn man verliert den Betriebs PTCH, wir haben einen Zellproliferationseffekt (und damit die Entwicklung von Zysten und Tumoren).
Standard radiologischen Aspekt:
Sehr oft asymptomatisch, die meisten Keratozysten während eines Standard-Röntgenuntersuchung entdeckt. Diese Prüfung in keinem Fall zu einer endgültigen Diagnose. Sie erlaubt eine Orientierung der Diagnose kann nur durch histopathologische Analyse bestätigt werden. Die häufigste Röntgen Aussehen odontogenen Keratozyste ist der Mono (in der Regel) oder polygéodes strahlendurchlässig, rund oder oval und mit dünnen Trennwände.
Im Oberkiefer kann Overlays mit Sinus oder Nasenhöhlen die Identifizierung dieser Funktionen sehr viel schwieriger zu machen. Wenn Keratozyste ist sehr umfangreich, Ausdünnung und kortikalen Knochen Verformung möglich ist.Es kann auf jedem halbOberKiefer zu entwickeln; die Kondylen werden immer eingehalten.
Letztere können auch wickeln Sie einen unentwickelten benachbarten Zahn (im Lieferumfang enthalten), ein überzähliger Zahn oder ein Odontom und Ursache der Verschiebung oder, sehr selten, die Wurzelresorption der anderen Zähne beim Kontakt entfernt. Peripheral edle Elemente (Nerven ...) nicht zerstört werden, sondern durch die Zyste zurückgesendet. Die Hauptdifferenz radiologische Diagnostik stellt sich mit Ameloblastom, follikulären Zysten dann einige.
4. Zurückgelegte Wegstrecke
Die Fälle von malignen Transformation odontogener Keratozysten in Plattenepithelkarzinome sind sehr selten. Nur 12 Fälle wurden in der Literatur berichtet. Diese Möglichkeit wird in Betracht gezogen, wenn es mehrere Wiederholungen, eine sehr aggressive Verhalten der Läsion sowie ein Ausfall sowohl der konservativen Behandlungen und aggressiven Behandlungen. Es wird oft bei älteren Patienten beobachtet. Fall ameloblastischen Transformationen sehr selten berichtet, sondern auch
. 5. Differenzialdiagnose
Die Differentialdiagnose der odontogenen Keratozyste muss mit drei anderen Zysten vor allem durchgeführt werden:
- Mit Ameloblastom:
This Zyste häufigsten männern bei und hat seine grösste Häufigkeit Vierten und in der Fünften Jahrzehnten. 80% der améloblastome Sind im Unterkiefer und insbesondere bei und DM Winkelpegel Ramus Entfernt. Kiefer Lokalisierungen Sind selten.
Ist aus Sicht Klinischer Ameloblastom Durch and a Hohe unbegrenztes Wachstum Rezidivrate gekennzeichnet. This Läsionen Heynckes oft Ein Volumen Genügend. Im Allgemeinen asymptomatisch, es wird nur Durch Zufall Einer zu Werden Röntgenuntersuchung entdeckt. Ansonsten Kann es Durch EINEN oder Durch infektiösen Folge Bewegungen und Zahnanomalien Evolution offenbart Werden.
Aus Sicht radiologischer Hut Eine Ameloblastom meist polygéodique Aspekt "Seifenblase" Grenzen und mit Regelmässige Inneren Trennwänden. Drusen Sind gut abgegrenzt, rund oder oval, in Verschiedenen Größen. Die edlen Elemente unterdrückt Werden, Nicht Zerstört. There is so viele Ähnlichkeiten with the Keratozyste , so that the Differentialdiagnose Nicht nur mit den Standard und Klinischen radiologischen Befunde. Die Differentialdiagnose Wird Durch Die DAHER Hauptsächlich Histologische Untersuchung, Sondern Auch vom CT-Scan.
- mit Einem follikulären Zyste:
This Zyste Männer im trafen häufiger Hut anzeigen Ihren Häufigkeitsgipfel in der Zweiten und Dritten Jahrzehnten.Sind Wesentlichen den im Dritten Unteren Molaren und den Oberen Eckzahn und der Zweiten Unteren Prämolaren betroffen.
Meist beschwerdefrei, haftet der follikulären Zyste eine den Hals und Eines Zahnes umgibt sterben Krone. Seine Entdeckung Wird am häufigsten während einer-Routine Röntgenuntersuchung oder Werden Durch Unerklärliche Zahnbewegung Motiviert.
Das ist das Röntgenbild Einer gut abgegrenzte Osteolyse, monogéodique und Rund um den Kranz oder Krone des Zahnes Entwickelt und Völlig Gebaut. Die Läsion groß werden can Recht und in den dringen aufsteigenden Ast der Unterkiefers. Es ist zu Schwieriger Erkennen, röntgenologisch einsetzen in den Hals Zahnes und sterben an DAHER Differenzierung Eines Keratozyste.
Daruber Hinaus ist es Möglich, EINEN der Wurzel Resorption benachbarten Läsion zu der Zähne beobachten; Welche seltener viel ist zum Keratozysten und ist der Zeichen der Beiden Hauptunterscheidungsradiologischen Entitäten.
- Mit Einem Follikularzyste:
Zyste This is a von vielzahl follikulären Zyste, Aber in Kontakt mit Zahnwurzeln Entwickelt, sterben das heisst, sterben Zähne Angebote sind nicht Vollständig ausgebildet. Seiner Entdeckung Kann Entweder Profilierung mittels Eines von Einem Röntgenbildes Motiviert verzögerten Ausbruch Eines Zahns Erfolgen; Entweder während Einer Infektions Folge (in der Regel im Prämolarenbereich). Andere Merkmale Sind mit Denen der Identisch follikulären Zyste.
- Mit Einem oder banalen entzündliche Zyste Zahn radiculo:
This Sind Die Läsionen häufigsten Kiefer. Treten Sie bevorzugt Bei Männern Oberkieferfront oder DM in der Ast der horizontalen Unterkiefers. Liegt Zwischen DM anzeigen Ihren Häufigkeitsgipfel Dritten und Vierten Jahrzehnten. Auf Ähnliche Läsionen zuvor gesehen, die ist in der Regel radikuläre Zyste asymptomatisch und Radiologische Routine Entdeckung . Es Kann Aber Durch Ausbeulen in Infektionsepisoden gekennzeichnet, Schwellung, NEBEN Einem Eiter Hohlen Zahn, nekrotischen oder Werden Nicht Vollständig Verarbeitet enthüllt. This Sind Zysten langsam Wachsende und Kann, in der Abwesenheit von Klinischen Zeichen, erweitern Deutlich.
Deren Röntgenbild ist das Eines monogéode, gerundet oder oval, in verbindung mit der Spitze Eines Durch Eine Zahns abgetötet und Grenze Knochenkondensation (sterben can bei Entzündungsattacken verschwinden) umgeben ist. Sie can im Fälle erheblicher Größe, ursache der Wurzelresorption benachbarten Wurzeln. 59
Die Differentialdiagnose Kann Auch Mit Anderen Zysten landen Wie odontogene Fibrom sterben, odontogene Myxom die, das einsame Zyste, oder der Knochen Angioma aneurysmatischen Zyste. 60 B.
B / Andere Veranstaltungen odontostomatologiques
Andere Dental odonto- Veranstaltungen Werden also available in der Basalzell-NAVUS-Syndrom, Aber mit Einer größeren Ungleichheit aufgetreten. Sind sie tatsächlich Nicht immer vorhanden ist, und wenn, Dann ist es mit Einer Frequenz wesentlich Niedriger Keratozysten als sterben, sterben Selbst Eines der Hauptkriterien der Krankheit. Sie sind oft in direktem zusammenhang mit der Entwicklung von und Kann als Keratozysten Einer der Ersten Rufzeichen stellen, um sie zu markieren (Diagnose und DAMIT von zur Basalzell-NAVUS Führen-Syndrom)
a / oder Einschlüsse Zahnfehlstellungen
Sie vor Allem sterben betreffen bleibenden Gebiss. Es ist Möglich, Eine von Persistenz Milchzähnen beobachten zu, Wenn Die letzte Zyste Zahn ist Durch Die sterben Wadenfänger Eruption verhindert beibehalten. Studien gezeigt have that in abhängigkeit von der Entwicklung der zeitlichen Zyste, sterben Folgen Zahnpegel Sind Unterschiedlich.In der Tat, Wenn Die Keratozyste Entwickelt Sich, Würde, ideal Wurzel Bau, verformung in den Wurzeln, oder Wie Krümmungen Biegungen Beobachtet Bajonett.
If you zu Einem Frühen in Gegenüberstellung Zahnelementbildung Entwickelt, das kann der Wurzel abreißen Führen.Durch Die nachteile, Wenn Die Zyste Wird nach DEM Zahndurchbruch Gebildet, so er can für die Reise und Verantwortlich Zahnfehlstellung Brust, Aber auf jeden fall Kann es Nicht für Wurzelresorption Zellstoff oder Abtötung Verantwortlich Brust.
b / Progenie
Schätzungsweise 25% der Fälle. Es can Eine wahre Entweder zu Einer pseudoprognathisme Prognathie oder ist oder Eine brachymaxillie gebunden Kombination von Beiden Brust. 61 Ihre Unterstützung Unterstützung Wird Durch und sein kieferchirurgische Kieferorthopädie. Progenie in Einem Patienten mit jungen-Gorlin-Syndrom (15)
c / Lippen- und Gaumenspalten
Die Lippen und / oder Werden Gaumenspalten in etwa 5-7% Fälle von der Basalzell-NAVUS-Syndrom gefunden. Eine Literaturrecherche von Lambrecht zeigt that von 719 Menschen sterben von-Gorlin-Syndrom, orofazialen Haben 61-Slot, 8,5% der Patienten. This can in Zwei Menschen Gruppen I Once, ob der Schlitz labial, alveolar und Gaumen (in 49 Gefallene) oder nur einteilen Spalt (13 Fälle)
Die Frequenz of this Ist nicht Mißbildungen dasselbe Zwischen der Patienten mit und Allgemeinen bevölkerung Basalzell NAVUS-Syndrom
.In der Tat, in der Allgemeinen bevölkerung, Ist das Auftreten von Lippen- Schlitze, alveolare Gaumen und that nur von 1/800 1/2500 und gegen Gaumenspalte Jeweils 1/1 und 14,7 / 55,3 der Patienten-Gorlin-Syndrom . Anomaly This can der Ersten Anzeichen Eines for the anwesenheit Eines Gorlin-Syndrom bei Einem innerhalb von Neugeborenen.
d ./ Parästhesien
Auch sie werden zur Entwicklung und Keratozysten Verbunden Treten auf, Wenn Die Zyste Ende erreicht Eine Größe Erhebliche, Wenn Es komprimiert oder EINEN verdrängt Nervenelement und verursacht Parästhesien. Es Kann Auch Eine entfernung Eines postoperative Komplikation der grossen Zyste mit den Grenzen der Nähe der in der Mutter Nerven .
e / Die améloblastome
Die améloblastome selten bei Patienten-Gorlin-Syndrom Auftritt, da nur Wenige Fälle gerechnet wurden in der Literatur berichtet Worden. zeigt den Fall Eslami 68 Jahre alten Eines Patienten mit dem-Gorlin-Syndrom Ende erreicht and a Ameloblastom.
Während für schnell alle Patienten von Waren dieser Krankheit betroffen, in Einem Alter Frühen erscheinen die Ersten Symptom, in der Regel Vor dem Alter von 35 jahren, die Ersten begannen Klinischen Anzeichen des Patienten zu viel Später erscheinen.
Es Seinen Ersten tatsächlich stellte Basalzellkarzinom im Alter von 50 jahren und Mehrere Keratozysten nur von 68 Jahren. Element interessant, wir Eslami beachten that der 3 4 Fälle von Basaliom-mole-Syndrom begann Ameloblastom in der Literatur Englischen aufgezeichnet ist, verknüpft, Ihre Ersten präsentierten Symptom Bildenden höheren Alter als in Einem sterben Mehrzahl von der Fälle Basalzell-NAVUS-Syndrom.
Das ist Durchschnittsalter für This Fälle 47,6 Jahre. This Entdeckung zeigt that mit Patienten Basalzell-NAVUS-Syndrom in Einem Alter Bildenden höheren Sind Die Entwicklung der Gefahr Ameloblastom.
Wie oben gesehen, Keratozysten und Sindh Ameloblastom, sowohl, gutartige Neubildungen mit Einer Hohen und große Rezidivrate Potenzial der Knochenzerstörung.
Allerdings ist und Infiltration Ameloblastom hartnäckiger, Eine noch radikalere chirurgische Resektion erfordern.
