DR R.J GORLIN , CHIRURGIEN-DENTISTE ET GENETICIEN , SON PARCOURS
CARACTERISTIQUES GENETIQUES DE LA NAEVOMATOSE BASOCELLULAIRE
A / GENERALITE
B / GENE IMPLIQUE DANS LA NAEVOMATOSE BASOCELLULAIRE
1. Généralités
2. Théorie du double événement
3. Voie de transduction Patched/sonic hedgehog
Acteurs de la voie de transduction patched/Sonic Hedgehog
Fonctionnement de la voie de transduction patched/Sonic Hedgehog
Voie de signalisation Patched/Sonic Hedgehog
Types de mutations et localisation
MANIFESTATIONS GENERALES, EN DETAIL , DE LA NAEVOMATOSE BASOCELLULAIRE
1 / MANIFESTATIONS DERMATOLOGIQUES
A / Naevi et carcinomes baso-cellulaires Généralités
B / Hyperkératoses palmo-plantaires Généralités
C / Autres manifestations dermatologiques
2 / MANIFESTATIONS SQUELETTIQUES
A / Calcifications des enveloppes du cerveau
B / Anomalies de forme du crane
C / Anomalies costales
D / Anomalies vertébrales
E / Anomalies des os de la main et du pied
F / Autres manifestations squelettiques
3 / MANIFESTATIONS OCULAIRES
A / Les malformations congénitales du globe
B / Les processus tumoraux du globe
4 / MANIFESTATIONS NEUROLOGIQUES
A / Méduloblastomes Statistiques
B / Autres manifestations neurologiques
5 / AUTRES MANIFESTATIONS GENERALES
A / Fibromes ovariens
B / Fibromes cardiaques
C / Hypogonadisme chez l’homme :
6 / MANIFESTATIONS ODONTOSTOMATOLOGIQUES
A / Les kératokystes odontogéniques
B / Autres manifestations odontostomatologiques
a / Inclusions ou malpositions dentaires
b / Prognathisme mandibulaire
c / Fentes labio-palatines
d / Paresthésies
e / L’améloblastome
QU EST CE QUE LE SYNDROME DE GORLIN ? DEFINITION
Le syndrome de Gorlin, aussi connu sous le nom de naevomatose basocellulaire (NBC), est une maladie héréditaire qui se caractérise par un ensemble d'anomalies du développement et par une prédisposition à développer différents cancers.
La prévalence est estimée entre 1/57 000 et 1/256 000, avec un ratio garçons/filles de 1:1. Les manifestations cliniques incluent la présence de nombreux carcinomes baso-cellulaires (CBC), de kératokystes odontogéniques des mâchoires, d'une hyperkératose palmo-plantaire, des anomalies du squelette, des calcifications ectopiques intracrâniennes et une dysmorphie faciale (macrocéphalie, fente palato-labiale et anomalies oculaires sévères). Un déficit intellectuel est observé chez 5% des patients. Les carcinomes baso-cellulaires (allant de papules ayant la couleur de la peau à des plaques ulcérantes avec un diamètre variant de 1 à 10 mm) sont habituellement localisés au niveau du visage, du dos et du thorax. Le nombre de carcinomes baso-cellulaires varie de quelques-uns à plusieurs milliers. Les anomalies squelettiques (affectant la forme des côtes, des vertèbres et du crâne) sont fréquentes. Peuvent également survenir des troubles oculaires, génito-urinaires et cardiovasculaires. Parmi les patients présentant le syndrome de Gorlin, 5-10% développent un médulloblastome, qui constitue une cause probable de décès précoce.
Le syndrome est dû à des mutations du gène PTCH1 et est transmis sur le mode autosomique dominant, à pénétrance complète et à expressivité variable. Le diagnostic clinique se base sur des critères spécifiques. Le diagnostic est confirmé par la recherche des mutations. Le conseil génétique est obligatoire. Le diagnostic anténatal est possible par échographie et par analyse ADN des cellules foetales, obtenues par amniocentèse ou par biopsie du trophoblaste. Le diagnostic différentiel principal inclut le syndrome de Bazex, le trichoépithélium multiple et le syndrome de Muir-Torre (voir ces termes). La prise en charge requiert une approche multidisciplinaire.
Les kératokystes sont éliminés par résection chirurgicale. Dans le cas des carcinomes basocellulaires, la chirurgie est indiquée lorsque le nombre de lésions est limité. Les autres traitements possibles sont l'ablation au laser, la thérapie photodynamique et une chimiothérapie locale.
La radiothérapie doit être évitée. Les analogues de la vitamine A pourraient jouer un rôle préventif contre le développement de nouveaux CBC. L'espérance de vie n'est pas significativement altérée mais des complications peuvent entraîner une morbidité non négligeable. Un suivi régulier par une équipe multidisciplinaire (dermatologues, neurologues et odontologues) est indispensable. Les patients doivent éviter toute exposition excessive aux rayons UV.
DR R.J GORLIN, CHRIRURGIEN- DENTISTE ET GENETICIEN , SON PARCOURS
Robert James Gorlin est né le 11 Janvier 1923 à Hudson, New York et décédé d’un lymphome le 29 Aout 2006 à Minneapolis, Minnesota à l’âge de 83 ans. Engagé dans l’armée au cours de la Seconde Guerre Mondiale, il a ensuite étudié la Dentisterie à l’Université de Washington où il a obtenu son diplôme en 1947. Il a poursuivi ses études en obtenant un Master de Chimie à l’Université d’Etat de l’Iowa. Après de nombreux postes universitaires, il obtient en 1956 un poste de Professeur et de Directeur de la Division de Pathologie Orale à l’Université du Minnesota où il restera jusqu’à sa mort .
Sa carrière prend un tournant en 1940 lorsque Gorlin découvre un livre de la dermatologiste Helen Curth traitant de l’achantosis nigricans. Cette expérience va contribuer à transformer Gorlin en « syndromologiste ». L’achantosis nigricans est une dermatose caractérisée par une hypertrophie papillaire végétante et une pigmentation localisée principalement aux aisselles, au cou et aux régions génito-crurales où la peau apparait rugueuse, épaissie et quadrillée. Cette affection dermatologique possède des formes malignes caractérisées par une association avec des adénocarcinomes gastriques. C’est donc une affection dermatologique qui a des répercussions sur d’autres systèmes de l’organisme. Cette découverte a captivé Gorlin qui s’est alors demandé si certaines maladies systémiques pouvaient présenter des manifestations orales. Gorlin est devenu internationalement connu pour son travail sur les manifestations orales de nombreux syndromes. Tout au long de sa carrière, Robert J. Gorlin et ses collègues ont étudié et nommé environ 100 syndromes et participé à l’isolation et la caractérisation des gènes responsables d’environ la moitié d’entre eux.
Le nom de Gorlin est devenu un nom commun, 7 familier pas seulement en génétique mais également en médecine. Cela a amené dermatologistes, généticiens et pathologistes à considérer Gorlin comme l’un d’entre eux. Robert J. Gorlin sera un écrivain très prolifique. Il a écrit environ 600 articles, 60 chapitres d’ouvrages et co-écrit ou édité 20 livres. Certains d’entre eux ont même été traduits en espagnol, en russe et en japonais. Le nom de Gorlin est devenu particulièrement indissociable de la naevomatose basocellulaire, d’ailleurs plus communément appelée syndrome de Gorlin.
En 1960, il décrit pour la première fois l’association de nombreux carcinomes basocellulaires cutanés, apparaissant sur l’ensemble du corps y compris sur des zones non exposées au soleil avec des déformations costales, des lésions osseuses ainsi que des défauts et des tumeurs oculaires. Il considère ces anomalies comme faisant parties d’un seul et même syndrome que l’on connaît aujourd’hui sous le terme de syndrome de Gorlin. Il aidera à la compréhension et la découverte du gène responsable de ce syndrome, le gène PTCH.
Lors de sa carrière à l’Université du Minnesota, ses nombreuses connaissances l’ont amené à donner des cours en pathologie, dermatologie, pédiatrie, obstétrique, gynécologie et l'otorhino-laryngologie. Il a aussi été Président de l'Association Internationale pour la Recherche Dentaire, de l'Académie Américaine de Pathologie Orale et de la Société Internationale de Biologie Cranio-faciale. Pour sa formidable contribution à la Science, la Médecine et la Société, Robert J. Gorlin a reçu de nombreuses et prestigieuses récompenses tout au long de sa carrière. Robert J. Gorlin est donc un dentiste de formation, qui par son travail a pu traverser les domaines de la Médecine et de la Génétique aussi bien que la Dentisterie.
CARACTERISTIQUES GENETIQUES DE LA NAEVOMATOSE BASOCELLULAIRE
A. GENERALITES
Bien avant R. J. Gorlin, cette entité clinique, connue sous le terme de naevomatose basocellulaire, a déjà été l’objet de descriptions, sous d’autres appellations. En 1894, Jarisch
décrit brièvement l’association de carcinomes basocellulaires avec des anomalies squelettiques chez un patient atteint de scoliose. Il ne fait aucune mention des autres
caractères de l’affection.
La même année, White a rapporté l’histoire familiale d’un patient porteur de carcinomes basocellulaires multiples, qui ont continué à apparaître tout au long de sa vie.
En 1932, Nomland décrit des carcinomes baso-cellulaires résultant de naevi pigmentés congénitaux.
En 1939, Straith est le tout premier à associer des kystes des maxillaires à des carcinomes basocellulaires cutanés multiples chez trois membres d’une même famille.
Il faudra patienter jusqu’en 1951 pour que deux dermatologistes, Binkley et Johnson, décrivent à la fois les épithéliomas cutanés ainsi que les kystes des maxillaires et les
modifications cérébrales.
C’est en 1960 à Minneapolis que Gorlin, chirurgien-dentiste et Goltz, dermatologiste réalisent la toute première description d’ensemble de cette entité clinique qu’ils considèrent comme un syndrome. Ils lui donneront le nom de naevomatose basocellulaire multiple avec kystes du maxillaire et côtes bifides.
Depuis lors, ce syndrome peut être désigné par plusieurs appellations telles que syndrome de Gorlin ou de Gorlin-Goltz, phacomatose de Gorlin-Goltz, syndrome des hamartomes basocellulaire…
Les plus anciens cas connus de naevomatose basocellulaire ont été révélés dans les années 1960. En effet, des kystes dentigères, des côtes bifides ainsi que d’autres stigmates du syndrome de Gorlin ont été trouvés sur deux squelettes masculins adultes de la collection de squelettes égyptiens de l’institut d’anthropologie de l’Université de Turin. Ces squelettes ont été exhumés à Assyut en Basse Egypte et datent de la période Dynastique. La période Dynastique commence par l'unification de l'Egypte, autrefois divisée en deux royaumes distincts, celui du Nord et celui du Sud et se situe autour de 3000 ans avant J-C.
La naevomatose baso-cellulaire est une maladie génétique héréditaire ayant un mode de transmission autosomique dominant. Cependant des cas sporadiques ont été rapportés : dans 60% des cas, aucun autre membre de la famille n’est atteint et 35 à 50% de ces cas représentent des néomutations. C’est pourquoi l’absence d’antécédents familiaux ne permet pas d’exclure le diagnostic de cette maladie. Elle est causée par la mutation du gène PTCH, localisé en 9q22.3-q31, qui est un gène
suppresseur de tumeur.
La naevomatose basocellulaire possède une forte pénétrance qui est la fréquence avec laquelle un gène manifeste ses effets. Elle est estimée à 97%. Cela signifie que 97% des
individus porteurs de ce gène sont atteints d’au moins une des manifestations de la maladie.
Cette maladie possède également une expressivité variable ; l’expressivité étant la qualité du gène de se manifester par des modalités variables quantitativement. La maladie se manifeste donc par de très nombreux symptômes, variables d’un patient à un autre. Il existe plus de quarante critères de diagnostic. Pour autant, chaque patient atteint de cette maladie ne présente pas l’ensemble des critères. Les critères de diagnostic ont été classés en deux catégories, majeure et mineure, en fonction
de leur fréquence d’apparition. Il est possible de poser un diagnostic de naevomatose basocellulaire lorsqu’un patient présente au minimum deux critères majeurs ou bien associe
un critère majeur à deux critères mineurs.
La prévalence est de 1/56000 avec un sex-ratio homme/femme de 3/1 . Selon Ghailan, la prévalence est de 1/60000 et hommes et femmes sont atteints dans les
mêmes proportions. Cette égalité d’atteinte entre les deux sexes s’explique fort bien par la transmission autosomique dominante de ce syndrome.
Selon Wolff, la prévalence oscille de 1/60000 à 1/120000 et les deux sexes sont affectés de la même manière. De plus le syndrome apparaît chez une grande variété de groupes
culturels et ne semble pas avoir de prédilection pour un type de peau en particulier.
La naevomatose basocellulaire reste une maladie rare puisque actuellement, seulement 700 cas ont été rapportés dans la littérature. Cependant le chirurgien dentiste se trouve en première position pour réaliser le diagnostic car les manifestations maxillo-dentaires font partie des premières manifestations de cette maladie.
B. GENE IMPLIQUE DANS LA NAEVOMATOSE BASOCELLULAIRE
1. Généralités
Le gène responsable de la naevomatose basocellulaire, le gène PTCH, fut découvert pour la première fois en 1996 par deux équipes de chercheurs. Sa découverte étant assez récente
l’ensemble des mécanismes génétiques menant à la maladie ne sont pas encore bien compris par la communauté scientifique, certains éléments restent encore flous.
Localisé sur le bras long du chromosome 9 et plus précisément en 9q22.3, ce gène est trouvé muté chez les patients porteurs du syndrome de Gorlin mais également dans les cas
sporadiques de carcinomes basocellulaires.
Ce gène joue un rôle important pour le développement embryonnaire et le contrôle de la prolifération cellulaire.
Dans un article de 1982, Fitzpatrick propose une théorie très différente des données actuelles afin d’expliquer l’origine de ce syndrome. Les techniques scientifiques de l’époque ne leur ont, en effet, pas permis de détecter l’anomalie génétique que l’on connaît aujourd’hui. Selon lui, ces anomalies multi-systémiques pouvaient s’expliquer pas l’existence d’un défaut enzymatique. La présence d’un tel défaut lors du premier trimestre de gestation pourrait expliquer les anomalies squelettiques ainsi que les calcifications des enveloppes du cerveau.
Plus tard, une anomalie dans le mécanisme enzymatique contrôlant l’épiderme pourrait mener à la formation des naevi, carcinomes baso-cellulaires, pits palmo-plantaires et des kératokystes odontogéniques. Cependant cette théorie se révèle insuffisante car elle ne permetpas de comprendre l’origine des tumeurs neurologiques telles que les médulloblastomes.
2. Théorie du double événement
L’étude de tumeurs dans la naevomatose basocellulaire a montré l’existence d’une perte de l’hétérozygotie pour la région 9q, suggérant que le gène PTCH pourrait être un gène suppresseur de tumeur. Cette notion d’anti-oncogène, dont l’archétype est le gène du rétinoblastome (Rb), repose sur la perte des deux allèles de ce gène dans une tumeur (perte de 13 l’hétérozygotie) d’après le modèle proposé par Knudson en 1971. En effet, selon sa « théorie des deux événements mutationnels », la première mutation serait responsable du syndrome malformatif et prédisposerait à la survenue de tumeurs et la deuxième entrainerait l’apparition des tumeurs.
Selon Cohen , afin qu’un gène suppresseur de tumeur soit inactivé, deux phénomènes sont nécessaires. Le premier est la mutation d’un allèle. Le second phénomène concerne la perte de l’autre allèle, connu sous le terme de perte d’hétérozygotie (ou LOH). Lorsque les deux allèles sont perdus, l’apparition de tumeurs s’effectue. Une perte d’hétérozygotie a été démontrée pour les carcinomes basocellulaires, kératokystes odontogéniques et médulloblastomes, qui sont trois des caractéristiques principales du syndrome de Gorlin.
Ainsi, dans les formes familiales du syndrome de Gorlin, la première des mutations pourrait être présente dès la conception et se trouver dans toutes les cellules de l’organisme. Ce premier événement mutationnel, générateur d’une prédisposition à développer une naevomatose basocellulaire dans la descendance, se transmet en dominance. Cet événement demeure latent et ne se démasque que si un second accident survient sur le chromosome
homologue au niveau du même locus, entraînant ainsi l’apparition du syndrome de Gorlin. Pour les cas familiaux, l’individu n’a besoin que d’une seule étape mutationnelle (puisqu’il a hérité de la première dans son matériel génétique). Par contre, l’apparition du syndrome de Gorlin chez un individu sans antécédents familiaux
nécessite la survenue de deux événements mutationnels successifs chez le même individu.
3. Voie de transduction Patched/sonic hedgehog
Le gène PTCH est l’homologue humain du gène patched chez la drosophile. Ce gène est un gène de segmentation de la drosophile (ou de polarité) intervenant dans la voie de
transduction patched/Sonic Hedgehog qui est impliquée dans le contrôle du développement embryonnaire et la prolifération cellulaire. Il fonctionne comme un régulateur du cycle cellulaire en stoppant la division cellulaire en l’absence de ligand et en l’activant dès que la liaison avec le ligand s’effectue.
Le syndrome de Gorlin provoque des atteintes au niveau du cerveau, des côtes, des vertèbres, des membres… Patched est exprimé dans l’ensemble de ces tissus.
Acteurs de la voie de transduction patched/Sonic Hedgehog :
Bien que de grands progrès aient été réalisés ces dernières années sur la compréhension du fonctionnement de la voie de signalisation patched/Sonic Hedgehog, de nombreuses
incertitudes devront encore être élucidées dans le futur.
- La protéine Patched ou phenylthiocarbamide, codée par le gène PTCH est une glycoprotéine composée de 1447 acides aminés avec douze domaines transmembranaires,
deux larges boucles extracellulaires requises pour la liaison du ligand SHH, une large boucle intra-cellulaire, un domaine amino-terminal intra-cellulaire et un domaine carboxy-terminal intra-cellulaire également.
On la retrouve mutée dans la naevomatose basocellulaire mais également dans les carcinomes basocellulaires et de rares cas de médulloblastomes.
La plupart des mutations sont des mutations non-sens qui aboutissent à la synthèse d’une protéine tronquée. (Lacombe) Gène Patched 1
- La protéine Sonic Hedgehog (SHH), codée par le gène shh localisé en 7q36. Cette protéine de 45kDa est clivée en deux domaines : un domaine amino-terminal de 20 kDa ayant une activité Zn hydrolase et un domaine carboxy-terminal de 25 kDa, autocatalytique, doué d’une
activité cholestérol transférase.
Le rôle du cholestérol dans cette voie de signalisation n’est pas tout à fait connu mais il pourrait jouer un rôle dans la limitation de la diffusion de la molécule SHH et la distribution
spatiale de ses effets.
- Enfin la protéine Smoothened (SMO), codée par le gène Smoothened localisé en 7q31-32.
Il s’agit d’une protéine de 115 kDa, composée de 787 acides aminés et appartenant à la famille des serpentines, récepteurs couplés à des protéines G (et qui présente une homologie avec la famille des récepteurs WNT). Cette dernière possède un domaine amino-terminal extra-cellulaire, un domaine carboxy-terminal intra-cellulaire et deux domaines (la troisième boucle intracellulaire et le septième domaine transmembranaire) impliqués dans la signalisation.
Fonctionnement de la voie de transduction patched/Sonic Hedgehog :
Patched a un rôle de récepteur pour le ligand SHH. En l’absence de SHH, SMO et Patched forment un complexe inactif. Patched agit comme un
régulateur négatif, un inhibiteur sur SMO.
Lorsque SHH vient se lier a Patched, ce dernier est alors inactivé et SMO est libre de transmettre le signal à l’intérieur de la cellule.
Plusieurs mécanismes ont été proposés pour expliquer l’activation de SMO et la transduction du signal :
- dans une première hypothèse, Patched et Smo forment un complexe. La liaison du ligand SHH à son récepteur Patched entraînerait un changement conformationnel de ce dernier et par conséquent une dissociation du complexe Patched/SMO. SMO serait alors libre de transmettre le signal à l’intérieur de la cellule.
- plus récemment, une seconde hypothèse propose une régulation non stoechiométrique par Patched. En effet, des analyses ont montré que la liaison de SHH entraînerait une diminutionde Patched et une augmentation de SMO dans la membrane cellulaire. La relation entre
atched et SMO serait donc non stoechiométrique, c’est à dire que Patched régulerait la 16
modification de SMO mais ne constituerait pas un réel complexe avec cette dernière (il n’y aurait pas d’interaction physique entre ces deux éléments).
SMO est donc activée et peut réaliser la transduction du signal vers le noyau.
La voie précise de transduction du signal via SMO n’est pas encore connue, cependant on sait que la cible finale est représentée par l’activation du facteur de transcription GLI (homologue de Cubitus interruptus ou Ci chez la drosophile). Dans les cellules non exposées à SHH, GLI forme un complexe tétramétrique avec d’autres protéines comme Costal-2 (COS2), Fused (FU) et le facteur suppresseur de Fused (SUFU). Sous cette forme, GLI peut être clivée en un fragment aminoterminal de 75 kDa qui peut transloquer vers le noyau et réprimer les gènes cibles dans ce dernier.
En présence du ligand SHH, le complexe se dissocie et GLI entier joue son rôle d’activateur transcriptionnel (passe dans le noyau et permet l’expression des gènes cibles).
Les gènes cibles sont :
- wingless (Wg) codant pour les membres de la famille WNT chez les vertébrés,
- décapentaplégic (dpp) codant pour les bone morphogénic protein (BMP) et les TFG béta
- ptc codant pour Patched lui-même.
Ces gènes sont essentiels pour un développement embryonnaire normal et la différentiation de certains tissus.
Le gène wingless code pour une famille de glycoprotéines impliquées dans des processus développementaux tels que la différentiation, la migration, la prolifération cellulaire et leur polarité. Un dérèglement de l’expression de ce gène est responsable d’anomalies développementales et d’oncogénèse.
Le gène décapentaplégic joue un petit rôle sur la transmission des informations permettant la différentiation dorso-ventrale de l’ectoderme embryonnaire.
Voie de signalisation Patched/Sonic Hedgehog
Rôle de Patched dans le développement :
a voie de signalisation Hedgehog joue un rôle important dans le développement des cellules souches dans une grande variété de tissus. PTCH est exprimé à la fois au cours du
développement embryonnaire et chez l’adulte, ce qui suggère qu’il continue d’exercer un rôle au cours de la période post-natale. Les modèles expérimentaux qui expriment de manière inappropriée un des composants de la voie de signalisation hedgehog révèlent d’importantes anomalies du développement. La ressemblance entre ces anomalies et celles caractéristiques du syndrome de Gorlin implique qu’une activation excessive de cette voie de signalisation peut être à l’origine des anomalies de développement et des tumeurs rencontrées dans lacnaevomatose baso-cellulaire.
Patched régule la transmission du signal délivré par SHH, signal impliqué dans plusieurs processus embryonaux tels que la formation du tube neural ventral, de parties du cerveau, des membres ou le développement dentaire...
Types de mutations et localisation :
Un grand nombre de mutations ont été rapportées par les auteurs. En effet, la naevomatose basocellulaire ne peut pas être attribuée à une seule et même mutation. Selon les patients, il existe des mutations non-sens, faux-sens, des inversions, des délétions… qui peuvent se répartir sur l’ensemble de la séquence codante du gène PTCH.
La majorité des mutations mènent à la création d’un codon stop qui entraine la synthèse d’une protéine tronquée.
Ces mutations se concentrent pour la majorité sur la large boucle intra-cellulaire, la large boucle extracellulaire et la région amino-terminale du gène PTCH.
Aucune corrélation n’a pu être établie entre la localisation de la mutation sur le gène et le phénotype observé chez les différents patients atteints du syndrome de Gorlin. Une des particularités de ce syndrome réside dans l’absence de corrélation génotype/phénotype. Pour des individus porteurs d’une même mutation, les tableaux cliniques peuvent se révéler très variables. Ceci suggère la possibilité de l’influence d’autres facteurs, comme des facteurs environnementaux ou génétiques, dans le développement de la maladie.
MANIFESTATIONS GENERALES , EN DETAIL , DE LA NAEVOMATOSE BASOCELLULAIRE
1 / MANIFESTATIONS DERMATOLOGIQUES
A / Naevi et carcinomes baso-cellulaires Généralités :
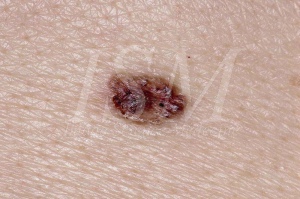
Les naevi basocellulaires constituent un des principaux signes de la naevomatose basocellulaires à tel point qu’en cas d’absence de ces naevi ; certains médecins éliminent d’emblé le diagnostic. Ils font partie des critères majeurs de la maladie, de la même manière que les kératokystes odontogéniques. Ils possèdent un grand potentiel évolutif de dégénérescence maligne en carcinomes basocellulaires.
On les retrouve dans plus de 90% des cas de syndrome de Gorlin. Il a été estimé qu’un patient sur 200 présentant un carcinome basocellulaire ou plus est atteint du syndrome de Gorlin. Plus le diagnostic de carcinome basocellulaire est réalisé tôt, plus cette proportion augmente. On considère qu’environ un patient sur cinq possédant un carcinome basocellulaire avant l’âge de 20 ans est atteint de naevomatose basocellulaire.
Age d’apparition :
Les carcinomes basocellulaires, dans le cadre du syndrome de Gorlin, apparaissent à un âge plus précoce que dans la population normale. Ils peuvent être présents dès la naissance ou se développer au cours de l’enfance. Le plus souvent, leur survenue commence à la puberté et dès l’âge de quarante ans, 90 % des patients touchés par cette maladie auront développé au moins un carcinome basocellulaire.
Nombre: Le nombre d’épithélioma basocellulaire est très variable. Même si l’on a déjà observé des cas clinique avec un seul épithélioma, dans la majorité des cas, leur nombre oscille entre une vingtaine et plusieurs milliers.
Localisation :
La localisation des ces tumeurs peut se faire tant sur les zones exposées au soleil (et donc aux rayons ultra-violets) que sur les zones non exposées. Les paupières, le nez, les joues et le front sont les sites les plus concernés mais le cou, le tronc et le creux axillaire sont également souvent touchés. Par contre, le cuir chevelu et les membres sont, la plupart du temps épargnés. Aspect clinique : Les naevi basocellulaires peuvent prendre des aspects cliniques variables.
Trois aspects sont généralement décrits :
- aspect évoquant le molluscum contagium qui est une affection cutanée contagieuse et inoculable dans certaines conditions. Elle est due à un virus et se caractérise par des petites élevures d’un blanc mat ou rosé de la grosseur d’une perle et dont le sommet est creusé par une dépression en ombilic.
- aspect de nodule hyperpigmenté de grande taille.
- aspect évoquant le carcinome basocellulaire classique. Cet aspect est le plus fréquent. Le naevus se présente alors comme une petite tumeur à surface lisse, translucide. Sa coloration est très variable, le plus souvent celle de la peau normale, parfois pâle rosé ou grisâtre. La consistance est beaucoup plus ferme que ne laisserait prévoir son aspect mais la base n’est pas infiltrée. La taille oscille entre 1 et 3 mm de diamètre. Si une de ces lésions augmente de taille ou commence à saigner ou à se recouvrir d’une croûte, une transformation maligne est à suspecter. Les carcinomes basocellulaires apparaissant dans le cadre de la naevomatose basocellulaire ne peuvent pas être clairement distingués de ceux apparaissant chez un patient non atteint par ce syndrome. Le seul élément caractéristique du syndrome de Gorlin est leur survenue en grand nombre et à un âge précoce ainsi que leur localisation, qui ne se fait pas forcement sur de zones exposées au soleil. L’aspect clinique de ces tumeurs est très variable allant de papules de couleur chair à brune ou à des plaques ulcératives. Ils peuvent être confondus avec des naevi ou des hémangiomes. Leur taille oscille entre 1 et 10 mm de diamètre.
Aspect histologique :
L’aspect histologique des naevi basocellulaires est celle d’une prolifération intra-dermique de cellules épithéliales, formant des plages bien délimitées, avec une assise périphérique souvent palissadique. Le stroma sous-jacent est riche en fibroblastes. L’aspect histologique des carcinomes basocellulaires de patients porteurs du syndrome de Gorlin n’est pas différent de ceux rencontrés chez les patients non-syndromiques. Lorsque le naevus se transforme en carcinome basocellulaire, on remarque des cellules plus volumineuses et plus claires, une augmentation du nombre de mitoses, un caractère inflammatoire de l’infiltrat. Il présente un stroma fibreux. La lésion peut devenir papullaire ou pédonculée. De profondes pénétrations, ulcérations ou invasions peuvent apparaître avec des infiltrations lymphocytaires. Ils peuvent être pigmentés à l’intérieur de la masse ou en périphérie.
Influence du soleil :
Une étude réalisée suggère que les patients porteurs d’une naevomatose basocellulaire présenteraient une sensibilité accrue aux rayons ultra-violets. Cela est appuyé par le fait que seulement 14% des patients atteints du syndrome de Gorlin dans le nord-ouest de l’Angleterre développent des carcinomes basocellulaires, contre 47% en Australie. Il insiste sur l’importance d’un diagnostic prénatal (lorsque l’un des parents est atteint) ou d’un diagnostic le plus précoce possible (pour les cas apparus de novo) afin de limiter l’exposition aux UV en cas de diagnostic positif.
Une autre étude, menée rapporte que 88% des carcinomes basocellulaire chez les femmes et 86% chez les hommes de la population générale apparaissent sur des zones corporelles exposées au soleil contre 59% chez les femmes et 65% chez les hommes porteurs de la naevomatose basocellulaire. Il conclut que la distribution anatomique des lésions suggère que le soleil n’est pas un facteur essentiel dans le développement des carcinomes basocellulaires. Leur présence en plus grand nombre dans les zones exposées suggère cependant que le soleil peut exacerber le développement des carcinomes basocellulaires et qu’il faut s’en protéger. Il ne reporte pas de corrélation significative entre le nombre de carcinomes basocellulaires et le nombre total d’heures d’exposition au soleil.
Seulement environ 40% des Afro-Américains avec un syndrome de Gorlin développent des carcinomes basocellulaires contre 90% des Caucasiens. L’étude menée chez 105 patients atteints du syndrome de Gorlin montre que seulement 38% des Afro-Américains présentent un ou plusieurs carcinomes basocellulaires contre 97% des Caucasiens. De plus, les Caucasiens développent des carcinomes basocellulaires de manière plus précoce. En effet, environ 50% des Caucasiens développent leur premier carcinome à 21,5 ans et 90% à 35 ans contre 20% et 40% aux mêmes âges chez les Afro-Américains
La plus petite fréquence de carcinomes basocellulaires chez le groupe des Afro-Américains peut être due à une meilleure protection contre les rayons ultra-violets du fait de la pigmentation de leur peau.
Influence des radiations ionisantes :
Les radiations ionisantes ont également été mises en cause dans le développement de ces tumeurs. En effet, les patients ayant subi des traitements par radiothérapie, présentent souvent de nombreux carcinomes basocellulaires dans les zones irradiées. Ils apparaissent en moyenne dans une période oscillant entre 6 mois et 3 ans après l’irradiation contre une moyenne de 21 ans chez un patient non porteur du syndrome de Gorlin.
Il faut noter qu’un des signes les plus précoces de la maladie est le médulloblastome. Or ce dernier est généralement traité par radiothérapie. Le risque de développer des carcinomes basocellulaires dans la zone irradiée est alors fortement augmenté. De plus, ces carcinomes sont beaucoup plus agressifs et donc plus difficiles à traiter. Pour ces raisons, il faut limiter autant que possible les traitements par radiothérapie et favoriser le traitement chirurgical.
Evolution :
Les carcinomes basocellulaires apparaissant dans le cadre du syndrome de Gorlin semblent être plus agressifs, plus difficiles à traiter. Ils peuvent aller jusqu’à entrainer le décès du patient par invasion (métastases au niveau du cerveau et des poumons majoritairement) ou par résistance aux traitements. La transformation maligne s’opère la plupart du temps après la puberté. Les tumeurs les plus agressives concernent le plus souvent les paupières et le nez et causent des destructions tissulaires pouvant mener à une importante défiguration des patients.
B / Hyperkératoses palmo-plantaires Généralités :
Ils sont également rencontrés sous les termes de « pits », trous ou puits palmo-plantaires. 26 Fréquence : Survenant à un âge précoce, leur présence peut être une aide importante pour le diagnostic précoce de la naevomatose basocellulaire puisqu’ils sont présents dans environ 65% à 80% des cas. Ils sont rencontrés avec à peu près la même fréquence dans tous les groupes d’âge. Hyperkératose palmaire après immersion des mains dans l’eau pendant quelques minutes.
Aspect clinique :
Ils correspondent à de petits trous ponctiformes qui sont des défauts de la couche cornée, qui est partiellement ou totalement absente. Rarement supérieurs à 1 ou 2 millimètres de diamètre et 1 millimètre de profondeur, leur fond peut être de couleur rose ou, si de la saleté s’est accumulée à l’intérieur, de couleur sombre. Ces lésions sont uniquement rencontrées sur les mains et les pieds et sont asymptomatiques. Lorsque le diagnostic clinique est difficile, le praticien peut faire immerger pendant quelques minutes les mains et/ou les pieds du patient dans de l’eau tiède. Les pits apparaîtront alors plus clairement.
Aspect histologique :
L’examen histologique d’un pits palmaire a montré un petit front prolifératif de cellules basales. Bien qu’il existe une désorganisation cellulaire à l’intérieur de ce front, une fissure entre ce front et le stroma adjacent n’a pas été observée. Une légère prolifération vasculaire et une fibrose ont été observées dans le derme adjacent. istologique
En conclusion, les pits palmo-plantaires sont une manifestation très caractéristique du syndrome de Gorlin. Ils sont une aide importante pour le diagnostic de ce syndrome du fait de leur importante fréquence d’apparition et l’âge précoce de leur survenue. Pour ces raisons, ils font partie des critères de diagnostic majeurs de la maladie, avec les carcinomes basocellulaires et les kératokystes odontogéniques.
La fréquence des kystes cutanés en rapport avec la naevomatose basocellulaire se situe entre 35 et 75 % selon les études. Les kystes épidermoïdes surviennent en général sur les zones pilleuses du corps et rarement sur les zones non pilleuses telles que la paume des mains et la plante des pieds. Cette localisation typique aux extrémités du corps serait une manifestation clinique distinctive de la naevomatose basocellulaire.
Cette description histologique est très similaire à celle des kératokystes odontogéniques, un des critères de diagnostic majeur de la naevomatose basocellulaire. Ces kératokyste cutanés, histologiquement proches des kératokystes des maxillaires, pourraient être une marque de fabrique typique de la naevomatose basocellulaire.
En conclusion, la découverte de kystes cutanés majoritairement localisés sur les mains et les pieds dont certains possèdent une forte ressemblance sur le plan histologique avec des kératokystes odontogéniques, doit être considérée comme une « perle clinique » pouvant nous mener vers un diagnostic de syndrome de Gorlin.
c / Autres manifestations dermatologiques
D’autres manifestations dermatologiques se retrouvent citées dans la littérature mais, du fait de leur très faible fréquence d’apparition, ne sont pas clairement décrites. Les patients atteints peuvent ainsi présenter des lipomes, des grains de milium, des taches « café au lait » ou encore des molluscums pendulums. Lipome situé au-dessus de la scapula droite. A noter également, la présence de nombreux naevi cutanés. Les molluscums pendulums sont des tumeurs fibreuses et flasques de la peau. Elles peuvent être planes et étalées, déprimées au palper, ou, au contraire, saillantes et même pédiculées. Les grains de milium sont une affection rare de la peau caractérisée par la formation de petites élevures brillantes, translucides, de la taille d’une tête d’épingle produite par la dégénérescence colloïde (transformation des cellules en une sorte de gelée) des couches superficielles du derme.
2 / MANIFESTATIONS SQUELETTIQUES
A / Calcifications des enveloppes du cerveau
Il est possible de rencontrer chez l’enfant des calcifications intra-crâniennes physiologiques ou pathologiques. Les premières sont très rares. Les secondes sont plus fréquentes et sont généralement décrites en association avec des tumeurs (telles que les médulloblastomes), des syndromes neuro-cutanés, des infections ou des désordres neuro-dégénératifs. Dans la population générale, les calcifications intra-crâniennes apparaissent en moyenne autour de 12 ans alors que, chez les patients porteurs du syndrome de Gorlin, elles sont visibles dès les premières années de la vie. Elles peuvent donc être utilisées comme un indicateur précoce pour le diagnostic de naevomatose basocellulaire. Elles concernent habituellement la faux du cerveau, qui est une partie de la dure-mère formant une cloison médiane entre deux hémisphères. Leur fréquence chez les patients porteurs du syndrome de Gorlin se révèle assez importante, puisqu’elle oscille entre 65 et 85 % selon les auteurs. Elles représentent le signe radiologique le plus fréquent. Des calcifications de la tente du cervelet sont également rencontrées mais beaucoup plus rarement que celle concernant la faux du cerveau. La tente du cervelet est la partie de la duremère comprise entre le cerveau et le cervelet. Il est également possible de rencontrer des agénésies du corps calleux mais cette anomalie reste rare.
B / Anomalies de forme du crane
Les patients atteints de naevomatose basocellulaire présentent des anomalies de la forme du crâne. Ces anomalies, bien qu’appartenant aux critères mineurs de la maladie, présentent un grand intérêt diagnostic puisqu’elles sont visibles dès la naissance. Chez certains patients le crâne est anormalement volumineux avec une proéminence des bosses frontales et pariéto-temporales. Les arcades sourcilières sont saillantes. L’hypertélorisme (écartement excessif des yeux) est fréquent et le prognathisme est parfois signalé. La base du nez est effondrée et élargie. Une déviation de la cloison nasale peut également être observée Selon une étude menée sur 105 patients porteurs du syndrome de Gorlin, une macrocéphalie est observée avec une plus grande fréquence (50% des cas) chez ces patients par rapport à la population normale. Il en est de même pour l’hypertélorisme qui est présent dans 42% des cas.
C / Anomalies costales
Bien qu’appartenant aux critères mineurs de la maladie, les anomalies costales sont présentent chez environ 42 % des patients atteints alors que leur fréquence dans la population est très faible. Elles sont donc largement présentes et sont principalement représentées par : - des bifidités, - des agénésies, - des synostoses costales. La synostose est une soudure totale de deux os voisins. Il est également possible d’observer des élargissements anormaux de l’extrémité antérieure ou postérieure des côtes. Les troisièmes, quatrièmes et cinquièmes côtes sont les plus fréquemment concernées par ces malformations même si toutes les côtes peuvent être atteintes. Certains auteurs comme Kimonis ont décidé de classer les anomalies costales parmi les critères majeures de la maladie pour deux raisons. La première est leur importante fréquence (42%) chez les patients atteints par rapport à la population générale. La seconde est que ces anomalies peuvent être facilement détectées à la naissance et représentent ainsi un élément de diagnostic majeur de la naevomatose basocellulaire dans la population pédiatrique. Si l’on applique les critères de Kimonis, un diagnostic plus précoce de la maladie peut être réalisé chez un grand nombre de patient. En effet, les autres critères majeurs de la maladie, tels que les kératokystes odontogéniques ou les carcinomes basocellulaires multiples ne sont pas présents dès la naissance ; ils apparaissent majoritairement au cours de l’enfance ou de l’adolescence. Les cliniciens doivent donc être très attentifs aux signes cliniques mineurs de la maladie au cours de la petite enfance. La présence d’un syndrome de Gorlin doit être recherchée chez les enfants qui naissent avec des anomalies costales, un fibrome cardiaque, une fente palatine ou labiale, une polydactylie ou une macrocéphalie.
D / Anomalies vertébrales
Selon une étude réalisée sur 105 patients porteurs du syndrome de Gorlin, des cas de scolioses ont été détectés de manière plus fréquente chez le groupe de personnes atteintes. Cependant, cette différence ne s’avère pas suffisamment importante pour être significative. Les individus malades semblent avoir des scolioses plus sévères que la population générale et le plus souvent, situées sur des sites d’anomalies développementales telles que des hémi-vertèbres ou des fusions des corps vertébraux. Ces anomalies développementales sont rencontrées dans 31 % des cas. Des cas de spina bifida ont été rapportés mais avec une fréquence peu différente de celle de la population générale. La spina bifida est une malformation consistant en une fissure de l’axe postérieur du rachis par défaut de soudure des points d’ossification sur une ou plusieurs vertèbres, à travers laquelle font hernie, sous forme d’une tumeur plus ou moins volumineuse, les méninges et parfois la moelle avec une quantité variable de liquide céphalorachidien. Les colonnes cervicales et thoraciques sont les plus concernées par cette malformation chez les patients atteints du syndrome de Gorlin
E / Anomalies des os de la main et du pied
La polydactylie n’est pas une malformation très fréquente puisqu’elle est rencontrée dans seulement 3% des cas, cependant elle est facilement détectable et présente dès la naissance. Son diagnostic est aisé. Les patients porteurs du syndrome de Gorlin peuvent également présenter un raccourcissement du quatrième et/ou du cinquième métacarpien. Ces malformations font partie des critères mineurs de la maladie car elles sont peu fréquentes.
F / Autres manifestations squelettiques
Déformation de Sprengel : 11% des personnes atteintes de naevomatose baso-cellulaire présentent une déformation de Sprengel ou élévation congénitale de la scapula. Cette anomalie consiste en un déplacement en haut et en dedans d’une ou des deux scapula avec déformation et parfois fixation au rachis de l’os déplacé. Cette élévation est due à un échec de la descente en position normale de cet os au cours de la vie fœtale.
3 / MANIFESTATIONS OCULAIRES
La fréquence des atteintes oculaires reste encore mal connue. Ceci est dû au fait qu’elles tiennent peu de place dans la description du syndrome de Gorlin, car la plupart des études publiées sur cette maladie traitent des critères majeurs. Les anomalies oculaires ne font l’objet que d’une mention et non d’une véritable description.
A / Les malformations congénitales du globe
Les principales malformations oculaires rencontrées sont la cataracte, le glaucome et le colobome dont les fréquences combinées sont estimées à moins de 14% selon Gorlin.
La cataracte est une affection oculaire aboutissant à l’opacité du cristallin ou à celle de sa capsule.
Le glaucome est une affection de l’œil caractérisée par une augmentation de la pression oculaire au dessus de 20 mm de mercure. Elle est due à une gêne à l’écoulement normal de l’humeur aqueuse.
Le colobome est une malformation du cristallin consistant en une encoche périphérique, unique ou multiple.
L’hypertélorisme, ou écartement excessif entre les deux yeux, est également fréquemment rencontré puisqu’il fait partie des caractéristiques faciales typiques de la naevomatose basocellulaire.
Outre ces anomalies, il est également possible de rencontrer des cas de strabisme, cataracte congénitale, microphtalmie ou de rétinite pigmentaire. La rétinite pigmentaire est un processus dégénératif de la rétine, bilatéral, familial et héréditaire. Il apparait dans l’enfance et est caractérisé par une baisse progressive de l’acuité visuelle et le rétrécissement du champ visuel, aboutissant tôt ou tard à la cécité
B / Les processus tumoraux du globe
Les paupières sont fréquemment le siège de naevi basocellulaires à risque de transformation maligne. Cette transformation peut se manifester par la constitution d’une ulcération torpide (c'est-à-dire qui n’évolue pas et ne régresse pas) et invasive ou par une infiltration en profondeur avec mutilation palpébrale. Les carcinomes basocellulaires des paupières sont en général plus agressifs et plus difficiles à traiter. De plus, leur traitement peut être à l’origine d’une importante mutilation faciale pour le patient.
Des cas de chalazion ont également été rapportés. Le chalazion est une petite tumeur palpébrale provenant de l’inflammation chronique d’une glande de Meibomius, glande sébacée située dans le cartilage tarse, constituant l’armature des paupières.
4 / MANIFESTATIONS NEUROLOGIQUES
A / Méduloblastomes Statistiques :
Dans la population générale, les médulloblastomes sont les tumeurs cérébrales les plus fréquentes au cours de l’enfance et concernent environ 20 % de la totalité de ces tumeurs. L’âge moyen du diagnostic est de 6 ans et 3 mois et les petits garçons sont atteints dans une proportion plus importante que les petites filles. Le traitement classique de ces tumeurs consiste en une exérèse chirurgicale associée à de la radiothérapie et de la chimiothérapie. Le taux de survie à 5 ans se situe entre 60 et 80 %. Environ 5 % des patients de moins de 5 ans ayant un médulloblastome sont atteints par le syndrome de Gorlin. Cette fréquence chute à 1 ou 2 % lorsqu’il s’agit d’adultes. L’âge moyen du diagnostic de médulloblastome chez les patients atteints est beaucoup plus précoce puisqu’il est de 2,1 ans.
Pour cette raison, il est très important de penser à rechercher la présence d’un médulloblastome dans les premières années de vie des enfants à risque de développer une naevomatose basocellulaire.
Les patients porteurs du syndrome ont un meilleur taux de survie que les patients non atteints.
Traitement :
L’exérèse chirurgicale, complétée par de la radiothérapie et/ou de la chimiothérapie est le traitement généralement proposé pour éradiquer les médulloblastomes. Dans les cas de syndrome de Gorlin, l’âge d’apparition de ces tumeurs est plus précoce ; or plus la radiothérapie est pratiquée tôt, plus le risque de séquelles à long terme (telles que des troubles de la croissance ou des dysfonctions endocriniennes) est important. De plus, au niveau des zones irradiées, le risque de développer des carcinomes basocellulaires ou des méningiomes est fortement augmenté. Ces carcinomes basocellulaires se révèlent beaucoup plus agressifs et plus résistant aux traitements.
Pour ces raisons, les traitements par radiothérapie chez les personnes porteuses du syndrome, doivent être évités autant que possible.
Les médulloblastomes répondant également aux 42 traitements par chimiothérapie, cette modalité de traitement doit être favorisée, au moins chez les patients les plus jeunes.
Certaines caractéristiques du syndrome de Gorlin, présentes à un âge précoce, comme le retard mental ou les calcifications de la faux du cerveau, peuvent être interprétées comme des manifestations uniquement causées par la tumeur. De plus, les signes les plus classiques de la maladie, comme les lésions cutanées ou les kératokystes des maxillaires, n’apparaissent que plus tard.
Pour ces raisons, la naevomatose basocellulaire peut être difficile à identifier à cet âge malgré la présence de la tumeur.
Certains auteurs pensent que le médulloblastome devrait devenir un des critères majeurs de la maladie car il fait partie, avec les altérations osseuses, des signes les plus précoces. Des investigations génétiques et des examens cliniques rigoureux doivent être proposés, à la recherche d’un éventuel syndrome de Gorlin, à tous les patients porteurs d’un médulloblastome avant l’âge de 3 ans ainsi qu’à leurs familles.
EN DETAIL
Médulloblastome
LE MÉDULLOBLASTOME
UNE TUMEUR DU SYSTÈME NERVEUX CENTRAL
EN DEUX MOTS...
Son origine
La classification internationale des cancers de l’enfant ( ICCC3 ) classe maintenant le médulloblastome parmi le groupe des tumeurs embryonnaires intracrâniennes, neuroectodermiques (PET - Primitive Neuroectodermal Tumor ).
Le médulloblastome se développe dans la partie inférieure du cerveau, dénommée fosse postérieure. La tumeur se développe à partir du quatrième ventricule ou du cervelet .
La tumeur dériverait d'une prolifération d'un progéniteur immature des neurones de la couche des grains .
Son épidémiologie
Le médulloblastome est la tumeur cérébrale maligne de la fosse postérieure la plus fréquente chez l’enfant. Il représente environ 20 % des tumeurs cérébrales, alors qu’il représente moins de 1 % des tumeurs cérébrales de l’adulte.
Sa prévalence est estimée à 1–9/100 000.
L'âge moyen de découverte est 5 ans. Il affecte plus souvent les garçons.
Les médulloblastomes se développent principalement aux dépens du vermis et du 4 ème ventricule.
LES FACTEURS DE RISQUE
LES FACTEURS ENVIRONNEMENTAUX
Un seul facteur a été identifié. Ce serait une exposition anténatale aux barbituriques. Cependant, cette hypothèse n’a pas été confirmée par toutes les études.
LES FACTEURS GÉNÉTIQUES
Les faits...
Une prédisposition génétique au médulloblastome est observée dans environ 10 % des cas. Si la tumeur se manifeste avant l’âge de 4 ans, la probabilité d'une prédisposition héréditaire est plus forte. Ces prédispositions correspondent essentiellement :
Le profil d’altérations génétiques est encore mal connu, bien que l'activation de certaines voies de transduction constitue un facteur pronostique reconnu.
Les techniques modernes ont montré que dans un tiers des médulloblastomes, la maladie était associée à la présence d’une anomalie chromosomique, l’isochromosome 17q. Cette anomalie est, pour certains auteurs, associée à un mauvais pronostic. De même, l'existence dans les médulloblastomes à grandes cellules et desmoplastiques d'une amplification du gène C-Myc serait associée avec un pronostic plus sombre.
LES SYMPTÔMES
Les premières manifestation de la maladie sont, le plus souvent des maux de tête surtout le matin et des vomissements traduisant une hypertension intracrânienne. Ces symptômes sont dus à l'existence d'une hydrocéphalie qui un excès de liquide céphalo-spinal (LCS) dans le cerveau.
Les symptômes suivants peuvent, aussi, être rencontrés :
Des problèmes visuels, comme une diplopie (vision double) intermittente ou permanente
Des problèmes d'écriture, une baisse de la performance scolaire
De l'indifférence, de l'apathie
Le délai entre le début des symptômes et le diagnostic, de 2 à 5 mois, est l’un des plus longs dans le cadre des cancers de l’enfant et s’explique par le manque de spécificité de ces symptômes et de leur attribution à des motifs psychologiques.
LE DIAGNOSTIC
LES MOYENS ?
C'est l'IRM qui est l'examen fondamental pour établir le diagnostic. Comme c'est une maladie pouvant donner des métastases dans le système nerveux, un bilan d'extension est réalisé qui comprend :
Une IRM complète de la moelle épinière
Une IRM post-opératoire pour étudier précisément l'existence d'un éventuel reliquat tumoral
Une recherche de cellules tumorales dans le LCS, 10 jours après l'opération
LA STADIFICATION
Les valeurs de T et de M
Au terme du bilan, le stade de la maladie peut être précisé. On distingue, dans une classification relativement ancienne, quatre valeurs pour la tumeur "T"
T1 < 3 cm
T2 > 3cm
T3 : T3a = atteinte du IV ème ventricule cérébral ; T3b = atteinte du plancher du IV ème ventricule ou du tronc cérébral
T4 = atteinte du III ème ventricule et/ou moelle épinière
Pour les métastases, on identifie 4 situations pour "M "
M0 = pas de métastase
M1 = présence de cellules tumorales dans le LCS
M2 = métastase(s) cérébrale(s)
M3 = métastase(s) médullaire(s)
M4 = métastase(s) systémiques(s)
Les groupes à risque
Ce bilan permet de classer le médulloblastome dans un des deux groupes suivants :
Le groupe à risque standard, correspond a T1, T2, M0, et sera traité par une exérèse complète de la tumeur
Le groupe à haut risque correspond aux autres cas
LES FORMES DE LA MALADIE
La plupart des médulloblastomes se développent dans le cervelet; cependant, ils peuvent émerger également dans d'autres parties du cerveau.
La classification de l'OMS distingue cinq types histologiques principaux :
La forme dite classique, 70 % des cas
La forme desmoplastique, de meilleur pronostic, 15 % des cas
La forme à nodularité extensive
La forme anaplastique
La forme dite à grandes cellules
Il existe maintenant une classification biomoléculaire :
Avec activation du WNT, 10 % des cas
Avec activation de la voie de signalisation SHH (Sonic Hedgehog) qui exerce une régulation majeure sur la prolifération des précurseurs des neurones à grains, 30 % des cas
Avec amplification de MYC (Thompson E ou 3), 25 % des cas
Avec gain de 17q (Thomson A et C ou 4), 35 % des cas
LE TRAITEMENT
Le pronostic de la maladie s'est beaucoup amélioré au cours des vingt dernières années. Actuellement, il existe une augmentation importante des taux de survie à long terme, atteignant jusqu’à 85 % dans les médulloblastomes de risque standard.
Les progrès des techniques chirurgicales, de la radiothérapie et le développement de la chimiothérapie contribuent à une meilleure prise en charge tant en termes de pronostic que de séquelles.
La prise en charge du traitement est multidisciplinaire et fait appel à la neuro-oncologie pédiatrique incluant le neurochirurgien, l'oncologue et le radiothérapeute. Le traitement fait appel à diverses modalités thérapeutiques :
La neurochirurgie traditionnelle ou stéréotaxique
La radiothérapie adjuvante comprenant une irradiation de la zone tumorale ainsi que du névraxe
La chimiothérapie en cas de métastases
Plus récemment la protonthérapie qui semble plus efficace et entraîner moins de séquelles
B / Autres manifestations neurologiques
Méningiomes :
Seulement 10 cas de naevomatoses basocellulaires associées à des méningiomes ont été rapportés dans la littérature. L’âge des patients oscille entre 18 et 64 ans. Il n’existe pas de prédominance sexuelle. Ces tumeurs semblent se développer dans les zones d’irradiation induites par le traitement d’un précédent médulloblastome.
Retard mental :
Des cas de retard mental et de difficultés à apprendre ont également été rapportés chez environ 5% des patients.
5 / AUTRES MANIFESTATIONS GENERALES
A / Fibromes ovariens :
Les jeunes filles et les femmes atteintes par le syndrome de Gorlin peuvent présenter des fibromes ovariens. Le fibrome est une des tumeurs bénignes des ovaires les plus fréquentes et représente environ 20% de la totalité des tumeurs ovariennes. Dans leur rapport initial de 1964, Gorlin et Goltz ont estimés que les fibromes ovariens étaient présents chez 75% des femmes atteintes par le syndrome de Gorlin.
Une série d’études menées plus récemment a montré une prévalence située entre 14 et 24%. L’âge moyen d’atteinte est de 30,6 ans mais peut aller de 16 à 45 ans. Il faut cependant noter que certaines de ces tumeurs ont été découvertes chez des patientes de moins de 3,5 ans. Pour cette raison, les patientes atteintes du syndrome doivent subir leur première échographie avant l’âge de 13 ans et avoir un suivi gynécologique régulier. Les fibromes ovariens croissent lentement et mesurent en moyenne 6 cm de diamètre même si certains peuvent aller jusqu’à 30 cm de diamètre.
Il a été reporté que les fibromes ovariens rencontrés dans le cadre du syndrome de Gorlin sont plus fréquemment bilatéraux et calcifiés que dans la population générale. Dans la majorité des cas, ces tumeurs sont totalement asymptomatiques. Elles n’interfèrent pas avec la fertilité des patientes et subissent rarement une transformation maligne. Le traitement de ces tumeurs est chirurgical.
B / Fibromes cardiaques :
Des cas de fibromes cardiaques ont été rapportés en rapport avec la naevomatose basocellulaire mais restent rares. Ils représentent 3 à 5% de l’ensemble des patients avec un fibrome cardiaque. Ils ne représentent que 5% des néoplasmes cardiaques. Ils apparaissent pendant l’enfance et dans 85% des cas chez des enfants de moins de 10 ans. Leur site de prédilection est le septum interventriculaire. Ces tumeurs sont à rechercher rapidement car elles apparaissent très tôt dans la vie et peuvent aller jusqu’à entrainer un décès précoce du patient.
C / Hypogonadisme chez l’homme :
Les hommes atteints par le syndrome de Gorlin peuvent présenter, très rarement, un hypogonadisme. L’hypogonadisme est l’état d’un sujet dont les glandes génitales ont une sécrétion interne insuffisante.
6 / MANIFESTATIONS ODONTOSTOMATOLOGIQUES
A. Les kératokystes odontogéniques
1. Généralités
Décrit pour la première fois par Philipsen en 1956, un kératokyste odontogénique est, selon l’Organisation Mondiale de la Santé ou OMS, « une tumeur bénigne uni- ou multi-loculaire, intra-osseuse d’origine odontogénique, avec une couche caractéristique d’épithélium pavimenteux stratifié parakératinisé et un potentiel pour un comportement agressif et infiltrant ». L’OMS recommande d’ailleurs le terme de « tumeur » car ceci reflète mieux sa nature néoplasique (comportement agressif, extension importante, tendance à la récidive…). Le terme de kyste primordial a longtemps été utilisé comme un synonyme du terme de kératokyste, entretenant une confusion. En effet, tous les kystes primordiaux ne sont pas des kératokystes dans la mesure où ils ne présentent pas tous une kératinisation. Inversement, tous les kératokystes ne sont pas des kystes primordiaux dans la mesure où ils ne se développent pas toujours en lieu et place d’une dent.
Le terme de kyste épidermoïde est, quant à lui, un synonyme. Ils représentent environ 11% de l’ensemble des kystes des maxillaires . Ils se situent au troisième rang, après les kystes radiculaires et folliculaires. Ils apparaissent trois fois plus souvent à la mandibule qu’au maxillaire.
Les kératokystes odontogéniques apparaissent dans tous les groupes d’âge, bien qu’un cumul de fréquence soit observé entre dix et quarante ans dans la population normale. Par contre, chez les patients porteurs du syndrome de Gorlin, le pic de fréquence est plus précoce et se situe donc dans la première décennie de la vie . Ces kystes constituent ainsi un des premiers symptômes de la maladie. Ceci confirme donc la place de choix de l’odontologiste dans le dépistage précoce de la naevomatose basocellulaire. Ils sont la plupart du temps découverts de manière fortuite lors d’une radiographie standard. Le pourcentage de patients asymptomatiques varie selon les études de 34 à 50%. Ils peuvent également être révélés par l’apparition de symptômes tels qu’une tuméfaction, des douleurs le plus souvent ; mais également un trismus, une cellulite, un retard d’éruption ou des malpositions dentaires entre autres…
Il faut noter que tous ces symptômes ne sont pas spécifiques des kératokystes odontogéniques. La présence de kératokyste odontogénique chez un jeune patient ou l’apparition de kératokystes récidivants ou multiples doit alerter le praticien et l’orienter vers un possible diagnostic de syndrome de Gorlin . Approximativement 5% des kératokystes sont associés à ce syndrome. Ils sont retrouvés chez 80% des patients avec un syndrome de Gorlin alors qu’ils ne sont présents que chez 5 à 7% de la population générale. Il existe plusieurs théories permettant d’expliquer l’étiologie des kératokystes odontogéniques.
Selon certaines les kératokystes odontogéniques se développeraient à partir des lames dentaires.
En effet, suite à la formation des germes dentaires, les lames dentaires se dissolvent dans les deux maxillaires. Au cours de ce phénomène, des îlots et des amas de cellules épithéliales peuvent persister dans le tissu conjonctif. Ce serait ces résidus épithéliaux, qui suite à une activation par des facteurs encore méconnus, seraient à l’origine de la formation des kératokystes. Selon d’autres, ils émaneraient du réticulum étoilé de l’organe de l’émail ou de la couche basale de l’épithélium de la muqueuse orale. En effet, cette autre théorie suppose que des ramifications de cette couche basale seraient à l’origine de la formation des kératokystes. Un des arguments qui appuie cette théorie est que l’on observe des récidives de ces kystes même lors de traitement chirurgical par résection des segments osseux affectés. Ceci suppose que l’origine de la récidive se situe en dehors de cet os et probablement dans les tissus mous adjacents. Il faut constater que ces deux théories ne sont nullement incompatibles puisque tant la lame dentaire que la muqueuse buccale sont à l’origine des tissus ectodermiques.
2. Caractéristiques
histologiques Le diagnostic de kératokyste odontogénique ne peut se faire que par l’analyse histolopathologique de celui-ci. L’examen radiologique permet, certes, d’orienter le praticien mais en aucun cas de faire un diagnostic définitif. Cet examen consiste en l’analyse du contenu et des parois kystiques obtenus soit après exérèse complète du kyste, soit après prélèvement d’un fragment de celui-ci au cours de son drainage ou de sa marsupialisation. Les caractéristiques histologiques principales reprises par la plupart des auteurs sont celles de Pinborg et Hansen en 1966 :
- un épithélium de revêtement malpighien habituellement très fin et uniforme en épaisseur, avec peu ou pas de papilles et possédant une forte activité mitotique ;
- une couche de cellules basales bien définie. Les cellules qui la composent sont de formes cubiques ou cylindriques et disposées le plus souvent selon un arrangement en palissade ;
- une fine couche, de quatre à huit assises cellulaires, de cellules épineuses présentant fréquemment un œdème intra-cellulaire ;
- la kératinisation est, le plus souvent, parakératosique (présence de noyaux). On la rencontre dans 83 à 97% des kératokystes. Parfois, il est possible de rencontrer une kératinisation de type orthokératosique (absence de noyaux) ou bien à la fois para- et orthokératosique. Pour certains auteurs comme Martin , le type orthokératosique devrait être individualisé car il serait moins agressif et moins récidivant que le type parakératosique ;
- la couche de kératine est souvent ondulée. La kératinisation seule n’est pas un critère spécifique du kératokyste odontogénique car d’autres kystes peuvent produire de la kératine.
- la paroi fibreuse du kyste est généralement fine et ne présente habituellement pas d’inflammation ;
- une fine coque conjonctive contenant souvent des ilots épithéliaux ou des kystes « filles » isolés en périphérie. Le contenu luminal du kyste possède un aspect liquide, jaune pâle ou crémeux, plus blanc, épais. Il existe une présence de kératine mais en quantité variable. En cas de réaction inflammatoire, on retrouve dans ce contenu des cristaux de cholestérol (se présentant sous la forme de paillettes brillantes), des corps hyalins, des polynucléaires et des amas bactériens. De plus, lorsque la paroi kystique est inflammée, l’épithélium s’épaissit et se transforme progressivement en un épithélium non kératinisé assez similaire à celui des kystes odontogènes inflammatoires banaux. La cytoponction est également un examen important pour le diagnostic du kératokyste odontogénique.
,En effet, la présence de kératine dans cette cytoponction est un élément qui aide au diagnostic (mais qui à elle seule ne suffit pas pour faire un diagnostic définitif car d’autres kystes tels que les kystes dentigères, primordiaux voire radiculaires ont un épithélium de surface qui peut être kératinisé).
Cependant, comme décrit ci-dessus, la présence de réaction inflammatoire induit la perte progressive de la parakératinisation des kératokystes et peut ainsi, parfois, empêcher un diagnostic cytologique correct. Un des autres éléments de la cytoponction permettant l’orientation vers un diagnostic de kératokyste est l’estimation du contenu protéique du kyste.
Lorsque ce contenu est mesuré à moins de 4 grammes de protéine pour 100 millilitres de liquide, le diagnostic de kératokyste est alors relativement sûr. Pour cette mesure, la présence d’une inflammation a également une mauvaise influence.
En effet, cette dernière a tendance à augmenter le contenu protéique et participe donc à la création de faux négatifs. Il existe également des méthodes visant à réaliser le diagnostic avant d’aborder la chirurgie. Ces techniques ne sont pas encore de pratique courante. La première est la recherche de l’antigène KAC (keratocyst antigene) dans le liquide du kyste. Cette présence confirmerait le diagnostic. La seconde serait la détermination immunohistochimique de l’expression de la cytokératine 10.
En effet, il s’agit d’un marqueur des épithéliums kératinisés. On la retrouve dans environ 50% des kératokystes odontogéniques mais également dans 10% des kystes radiculaires et 3% des kystes dentigères. Cependant l’étude de l’expression de cette cytokératine ne serait pas perspicace au sein des kératokystes car ces lésions semblent en perdre l’expression après décompression.
3. Caractéristiques cliniques et radiologiques
Localisation :
Le kératokyste odontogénique siège plus souvent dans la mandibule que dans le maxillaire. En effet, on le rencontre trois fois plus souvent dans la mandibule, et plus particulièrement au niveau de l’angle et du ramus, suivi des localisations dans la région des premières et deuxièmes molaires et des localisations plus antérieures.
Au niveau maxillaire le siège du kératokyste est un peu plus antérieur, dans la région prémolomolaire, puis la région canine et exceptionnellement la région antérieure.
Age d’apparition : Les kératokystes odontogéniques associés au syndrome de Gorlin se développent, de manière générale, durant la première décennie de la vie et atteignent leur pic d’incidence entre 20 et 30 ans, contrairement aux kératokystes non associés à ce syndrome et qui sont découverts plus tardivement, au cours de la quatrième décennie (âge moyen 37 ans).
Du fait de ce développement précoce, ils constituent un des premiers symptômes perceptibles de la maladie et confirment la place de choix de l’odontologiste dans le diagnostic de ce syndrome.
Taille :
Leur volume est des plus variable, allant de quelques millimètres à un envahissement d’une hémi-mandibule (18). Du fait qu’ils sont le plus souvent découverts tardivement, lors d’un examen radiologique de routine, il n’est pas rare qu’ils aient déjà atteint un volume considérable. Leur longueur moyenne est de 5 centimètres. (34)
Sex ratio :
Dans la population normale les hommes sont affectés plus souvent que les femmes avec un sex ratio de 2/1. Par contre chez les patients atteints de naevomatose basocellulaire, cette différence entre les sexes n’existe pas et il semblerait même que les femmes soient affectées un peu plus souvent que les hommes. (5) Symptômes : La majorité des kératokystes odontogéniques est exempt de tout symptôme. En effet, 34 à 50% seraient asymptomatiques et donc découverts seulement à l’occasion d’un examen dentaire ou radiologique de routine. Lorsque les symptômes existent, ils se manifestent le plus souvent par l’apparition d’une voussure ou tuméfaction osseuse progressive pouvant être douloureuse.
Les phénomènes inflammatoires ne sont alors pas rares et se caractérisent par la survenue d’abcès, de cellulites voire de fistulisations accompagnés parfois d’un trismus. En dehors de ces phénomènes, le kératokyste peut être découvert du fait des malpositions, des malocclusions dentaires ou des retards d’éruption qu’il peut provoquer et qui vont interpeller le praticien.
Il faut cependant noter que toutes ces manifestations ne sont pas du tout spécifiques aux kératokystes odontogéniques et nécessiteront un examen radiologique et surtout histopathologique afin de pouvoir confirmer le diagnostic.
Croissance :
Une des caractéristiques cliniques principales des kératokystes odontogéniques est leur fort potentiel de croissance. Ceci représente même un des critères ayant amené l’OMS à les considérer comme des néoplasmes bénins. Différentes théories sont avancées afin d’expliquer cette caractéristique :
- une première explication serait l’existence d’un enzyme collagénase à l’intérieur du kératokyste. Elle serait responsable d’une activité collagénolytique qui expliquerait ce fort potentiel de croissance ;
- une autre théorie serait l’existence d’une résorption osseuse induite par des prostaglandines E et F sécrétées par le kyste ;
- enfin, une plus grande osmolarité du fluide kystique ainsi qu’un pouvoir mitotique augmenté au niveau de la paroi kystique pourraient être également responsable de cette forte croissance. L’extension de ces kystes se fait au sein de l’os en remplaçant l’os spongieux plutôt qu’en s’étendant latéralement comme les autres kystes vers les régions corticales et périostées.
Lorsque les kératokystes sont volumineux, ils se moulent sur les bords de l’os et des dents, la 53 corticale (externe le plus souvent) est alors amincie, parfois même perforée. Des cas de fractures mandibulaires et/ou de déformations faciales importantes ont été rapportés mais restent cependant très rares. Les éléments nobles tels que le paquet vasculo-nerveux du canal alvéolaire inférieur sont, quant à eux, toujours respectés mais peuvent être refoulés (et entraîner très rarement une paresthésie).
Récurrence, mécanisme de la récurrence :
Les kératokystes odontogéniques ont également un taux de récurrence très élevé. Cela représente une de leur principale caractéristique clinique. Ce taux est encore plus important chez les patients porteurs de naevomatose basocellulaire et avoisine les 82% contre 25 à 60% chez les patients sains (33). Il existe trois mécanismes expliquant les raisons de ce fort taux de récidive :
- l’exérèse incomplète des pourtours du kyste, soit du fait d’une paroi kystique très mince et friable, soit du fait d’une perforation de la corticale osseuse avec adhérence du kyste aux tissus mous adjacents ;
- l’apparition d’un kératokyste odontogénique à partir d’un kyste satellite, d’un microkyste ou de restes odontogéniques oubliés après la chirurgie ;
- le développement d’un nouveau kératokyste odontogénique dans une aire adjacente et qui est interprété comme une récurrence alors que c’est une formation de novo n’ayant pas de rapport avec la lésion originelle. Selon d’autres auteurs , ce taux de récidive est également influencé par :
- la taille, la localisation (ramus), la morphologie de la lésion (en particulier multiloculaire) qui rendent le traitement et la chirurgie plus difficile
- le choix de la technique chirurgicale (conservatrice ou agressive) La plupart des récidives sont constatées dans les 5 à 7 ans suivant la première prise en charge.
Cependant des cas ont été rapportés plus de 10 ans après le traitement. 25% des cas de récidives se situent à partir de 9 ans et plus. 54 Un aspect essentiel du traitement des kératokystes odontogéniques est donc un suivi au long terme (une dizaine d’années semble idéal pour pouvoir diagnostiquer la quasi-totalité des récidives à un stade précoce). Certaines caractéristiques des kératokystes odontogéniques ont amenées l’OMS à les reclasser sous le terme de tumeurs ou néoplasmes bénins plutôt que kystes : - tout d’abord leur comportement : ces kystes sont localement très destructeurs, invasifs et possèdent un fort taux de récidive ; - ensuite leur histopathologie : les études ont montré que leur couche basale bourgeonne dans le tissu conjonctif. De plus l’OMS a noté que des figures de mitoses sont fréquemment trouvées dans la couche supra-basale ; l’épithélium des kératokystes a un index mitotique augmenté par rapport aux autres kystes des maxillaires (mais similaire à l’améloblastome) ; - pour finir l’aspect génétique : le gène PTCH est un gène suppresseur de tumeur localisé sur le chromosome 9q22.3-q31. Ce gène est à la fois responsable de la naevomatose basocellulaire mais aussi responsable de la formation de kératokystes odontogéniques isolés. En temps normal PTCH code pour une protéine signal transmembranaire qui forme un récepteur avec l’oncogène SMO (smoothened) pour le ligand SHH (sonic hedgehog). Lorsque SHH active ce récepteur il y a une inhibition du signal de croissance de différents tissus et de prolifération. Il intervient également dans la régulation du métabolisme lipidique. Par contre si l’on perd le fonctionnement de PTCH, on aura un effet de prolifération cellulaire (et donc développement de kystes et tumeurs).
Aspect radiologique standard :
Très souvent asymptomatiques, la plupart des kératokystes sont découverts à l’occasion d’un examen radiologique standard. Cet examen ne permet en aucun cas de poser un diagnostic définitif. Il permet une orientation du diagnostic qui ne pourra être confirmée que par l’analyse histopathologique. L’apparence radiologique la plus fréquente d’un kératokyste odontogénique est celle de mono (le plus souvent) ou polygéodes radioclaires, rondes ou ovales et avec de fines cloisons de refend.
Au niveau du maxillaire, des superpositions avec les sinus ou les cavités nasales peuvent rendre le repérage de ces caractéristiques beaucoup plus difficile. Lorsque le kératokyste est très étendu, un amincissement et une déformation des corticales osseuses est possible. Il peut se développer sur tout un hémi-maxillaire ; les condyles sont toujours respectés.
Ce dernier peut également envelopper une dent adjacente non évoluée (incluse), une dent surnuméraire ou encore un odontome et entraîner le déplacement voire, très rarement, la rhizalyse des autres dents situées à son contact. Les éléments nobles périphériques (nerfs…) ne sont pas détruits par le kyste mais refoulés. Le principal diagnostic radiologique différentiel se pose avec l’améloblastome, puis certains kystes dentigères.
4. Evolution
Les cas de transformation maligne des kératokystes odontogéniques en carcinomes épidermoïdes sont très rares. Seulement 12 cas ont été rapportés dans la littérature jusqu’à aujourd’hui. Cette possibilité est à envisager lorsqu’il existe de multiples récurrences, un comportement très agressif de la lésion ainsi qu’un échec à la fois des traitements conservateurs et des traitements agressifs. Elle est souvent observée chez des patients âgés. Des cas de transformations améloblastiques sont rapportés mais également très rares
. 5. Diagnostique différentiel
Le diagnostic différentiel du kératokyste odontogénique doit se faire avec trois autres kystes principalement :
-avec un améloblastome :
Ce kyste est plus fréquemment rencontré chez le sexe masculin et possède son pic d’incidence dans les quatrième et cinquième décennies. 80% des améloblastomes se situent à la mandibule et plus particulièrement au niveau de l’angle et du ramus. Les localisations maxillaires sont plus rares.
D’un point de vue clinique, l’améloblastome est caractérisé par une croissance illimitée et un fort taux de récidive. Ces lésions ont souvent un volume assez important. Généralement asymptomatique, il ne sera découvert qu’au hasard d’un examen radiographique. Dans le cas contraire, il peut être révélé par un épisode infectieux ou par des déplacements et anomalies d’évolution dentaires.
D’un point de vue radiologique, l’améloblastome a le plus souvent un aspect polygéodique en « bulle de savon », à limites régulières et avec des cloisons de refend. Les géodes sont bien délimitées, rondes ou ovalaires, de dimensions variables. Les éléments nobles sont refoulés et non détruits. Il existe donc beaucoup de similarités avec le kératokyste, rendant le diagnostic différentiel impossible à réaliser avec les seules observations cliniques et radiologiques standard. Ce diagnostic différentiel se fera donc essentiellement par l’examen histologique, mais également par l’examen tomodensitométrique.
- avec un kyste dentigère :
Ce kyste rencontré plus fréquemment chez l’homme possède son pic d’incidence dans les deuxième et troisième décennies. Il intéresse essentiellement la troisième molaire inférieure, puis la canine supérieure et la deuxième prémolaire inférieure.
La plupart du temps exempt de tout symptôme, le kyste dentigère adhère au collet d’une dent incluse et en entoure la couronne. Sa découverte se fait le plus souvent au cours d’un examen radiologique de routine ou peut être motivée par des déplacements dentaires inexpliqués.
L’aspect radiologique est celui d’une ostéolyse bien limitée, monogéodique et développée autour de la couronne ou du collet d’une dent incluse et totalement édifiée. La lésion peut devenir assez volumineuse et envahir la branche montante de la mandibule. Il est alors plus difficile de percevoir radiologiquement l’insertion de celle-ci au collet de la dent incluse et donc de la différentier d’un kératokyste.
De plus, il est possible d’observer une rhizalyse des dents adjacentes à la lésion ; ce qui est beaucoup plus rare pour les kératokystes odontogéniques et constitue le signe radiologique distinctif principal de ces deux entités.
- avec un kyste folliculaire :
Ce kyste est une variété de kyste dentigère mais qui se développe au contact de follicules dentaires ; c’est-à-dire de dents incomplètement formées. Sa découverte peut se faire soit au moyen d’un cliché radiographique motivé par un retard d’éruption d’une dent permanente ; soit à l’occasion d’un épisode infectieux (le plus souvent dans la région prémolaire mandibulaire). Ses autres caractéristiques sont identiques à celles du kyste dentigère.
- avec un kyste inflammatoire banal ou radiculo-dentaire :
Ces lésions sont les plus fréquentes des maxillaires. Elles apparaissent préférentiellement chez le sexe masculin dans le secteur antérieur des maxillaires ou la branche horizontale de la mandibule. Leur pic de fréquence est situé entre les troisième et quatrième décennies. De la même manière que les lésions vues précédemment, le kyste radiculaire est le plus souvent asymptomatique et de découverte radiologique de routine. Il peut cependant être révélé lors d’épisodes infectieux caractérisés par une voussure, une tuméfaction, une suppuration située en regard d’une dent cariée, nécrosée ou incomplètement traitée. Ces kystes sont d’évolution lente et peuvent, en l’absence de signes cliniques, s’étendre considérablement.
Leur aspect radiologique est celui d’une monogéode, arrondie ou ovalaire, en rapport avec l’apex d’une dent mortifiée et entouré d’un liseré de condensation osseuse (qui peut disparaître lors des poussées inflammatoires). Ils peuvent, en cas de taille importante, entraîner la rhizalyse des racines adjacentes. 59
Le diagnostic différentiel peut également se poser avec d’autres kystes tels que le fibrome odontogénique, le myxome odontogénique, le kyste solitaire, l’angiome osseux ou encore le kyste anévrysmal. 60 B.
B / Autres manifestations odontostomatologiques
Les autres manifestations odonto-stomatologiques sont également rencontrées dans la naevomatose basocellulaire mais avec une plus grande disparité. En effet, elles ne sont pas présentes systématiquement et, lorsqu’elles le sont, c’est avec une fréquence beaucoup moins importante que les kératokystes odontogéniques qui constituent, eux, un des critères majeurs de la maladie. Elles sont souvent directement liées au développement des kératokystes et peuvent représenter un des premiers signes d’appels permettant de les mettre en évidence (et donc mener à l’établissement du diagnostic de naevomatose basocellulaire)
a / Inclusions ou malpositions dentaires
Elles concernent surtout la denture permanente. Il est possible d’observer une persistance de dents lactéales lorsque la dent définitive est retenue par le kyste, qui empêche son éruption. Des études ont mis en évidence que, selon le moment du développement du kyste, les conséquences au niveau dentaire sont différentes. En effet, lorsque le kératokyste se développe lors de l’édification radiculaire, on observe des déformations au niveau des racines telles que des incurvations ou courbures en baïonnette.
S’il se développe précocement, en juxtaposition à un élément dentaire en formation, il peut causer la dilacération des racines. Par contre, si le kyste se forme après l’éruption dentaire, il peut être responsable de déplacements et malpositions dentaires mais en aucun cas il ne peut être responsable de rhizalyse ou de mortification pulpaire.
b / Prognathisme mandibulaire
Il est estimé à 25% des cas. Il peut s’agir soit d’un vrai prognathisme, soit d’un pseudoprognathisme lié à une brachymaxillie, soit d’une association des deux. 61 Leur prise en charge se fera par chirurgie orthognathique et orthodontie. Prognatisme mandibulaire chez un jeune patient atteint par le syndrôme de Gorlin (15)
c / Fentes labio-palatines
Les fentes labiales et/ou palatines sont rencontrées dans environ 5 à 7 % des cas de naevomatose basocellulaire. Une étude de la littérature menée par Lambrecht montre que sur 719 personnes affectées par le syndrome de Gorlin, 61 possèdent une fente orofaciale, soit 8,5% des patients. Ces personnes peuvent être séparées en deux groupes selon que la fente soit labiale, alvéolaire et palatine (pour 49 cas) ou uniquement palatine (pour 13 cas)
La fréquence de ces malformations n’est pas la même entre la population générale et les patients porteurs de naevomatose basocellulaire
. En effet, dans la population générale, la fréquence des fentes labiale, alvéolaire et palatine est de 1/800 et celle des fentes uniquement palatines de 1/2500 contre respectivement 1/14,7 et 1/55,3 chez les patients atteints du syndrome de Gorlin. Cette malformation peut être une des premières indications de la présence d’un syndrome de Gorlin chez un nouveau-né.
d ./ Paresthésies
Elles sont également liées au développement des kératokystes et se rencontrent lorsque le kyste atteint une taille si importante qu’il comprime ou refoule un élément nerveux et entraine la paresthésie. Elle peut être également une complication post-opératoire de l’exérèse d’un kyste volumineux dont les limites sont proches du nerf.
e / L’améloblastome
Les améloblastomes sont rarement rencontrés chez les patients porteurs du syndrome de Gorlin puisque peu de cas ont été rapportés dans la littérature. Eslami présente le cas d’une patiente de 68 ans atteinte par le syndrome de Gorlin et présentant un améloblastome.
Alors que pour la quasi-totalité des patients touchés par cette maladie, les premiers symptômes se manifestent à un âge précoce, généralement avant 35 ans, les premiers signes cliniques de cette patiente ont commencé à apparaître beaucoup plus tard.
En effet, elle a présenté son premier carcinome basocellulaire à l’âge de 50 ans ainsi que des kératokystes multiples à partir de 68 ans seulement. Elément intéressant, Eslami nous fait remarquer que 3 des 4 cas de naevomatose basocellulaire associés à un améloblastome enregistrés dans la littérature anglaise, ont également présenté leurs premiers symptômes à un âge plus avancé que pour la majorité des cas de naevomatose basocellulaire.
L’âge moyen pour ces cas est de 47,6 ans. Cette découverte indique que les patients présentant une naevomatose basocellulaire à un âge plus avancé seraient plus à risque de développer des améloblastomes.
Comme vu précédemment, les kératokystes odontogéniques et les améloblastomes sont, tous les deux, des néoplasmes bénins avec un fort taux de récurrence et un grand potentiel de destruction osseuse.
Cependant, l’améloblastome est plus persistant et encore plus infiltrant, ce qui nécessitera une exérèse chirurgicale encore plus radicale.
Notes aux visiteurs, Notes for visitors, Note per i visitatori , Notas para los visitantes, Hinweise für Besucher, Примечания для посетителей, 遊客注意事項,Notes voor bezoekers, Ziyaretçiler için notlar
Étant seule à créer ce site , sans l'aide de quiconque , si vous voyez des oublies, ou des erreurs sur mon site, merci de m 'en informer par mail .. si vous voulez apporter des temoignages, (bien entendu de maniere anonyme, des renseignements complementaire , photos , etc , contacter moi . je me ferai un plaisir de les publier ..
.
D'autre part, pour faciliter la lecture de chacun, en langue étrangère, j'ai utilisé plusieurs couleurs d'écriture suivant les langues traduitent dans la page d'accueil . Dans les autres chapitres suivez les menus déroulants pour accéder aux traductions . :
le NOIR = francais
BLEU = anglais
VERT = italien
ESPAGNOL = violet
ALLEMAND = orange
MARRON = russe
ROUGE = chinois (traditionnel )
JAUNES FONCé = neerlandais
langues supplementaires :
turc
Utilisant google traduction , si les traductions sont erronées, merci de m'en informé également
je vous remercie d'avance .
Corinne
Beingalonein creatingthis site withoutthe help of anyone, if you seeforgotten, or errorson my site,thank youmto informby email.. ifyou want to maketestimonies, (of courseanonymousway,thesupplementaryinformation,photos,etc.,contactme.Iwould be happyto publish them.. . Moreover, to facilitate the reading of each foreign language, I have used several writing colors following languages traduitent in the home page. In the other chapters follow the drop down menus to access translations
BLACK=french BLUE =English GREEN =Italian SPANISH=purple GERMAN = orange BROWN =Russian RED =Chinese (Traditional)
Dutch = dark yellow
Using googletranslation,if the translationsare wrong, thank you alsoinformed me
I thank you in advance .
Corinne
Essere solinella creazione diquestosito, senza l'aiutodi nessuno, sesi vededimenticato, oerrori sulmio sito, graziemdi informarevia e-mail.. sesi vuole farele testimonianze, (di modoovviamenteanonima, le informazioni supplementari, foto, ecc,in contatto con me. sarei felicedi pubblicarle.. . Inoltre, per facilitare la lettura di ogni lingua straniera, ho usato diversi colori di scrittura seguenti lingue traduitent nella home page. Negli altri capitoli seguire il menu a discesa per le traduzioni di accesso
Utilizzando traduzione di Google, se le traduzioni sono sbagliate, grazie che mi haianche informato
Vi ringrazio in anticipo.
Corinne
Estar soloen la creación deeste sitiosin la ayuda denadie,si vesolvidado,oerroresen mi sitio, gracias mde informarporcorreo electrónico.. siquieres hacertestimonios,(de maneraanónimacurso,la información complementaria, fotos, etc., póngase en contacto conmigo. yo sería felizpublicarlas.. .
Por otra parte, para facilitar la lectura de cada idioma extranjero, he utilizado varios colores que escriben los siguientes idiomas traduitent en la página principal. En los otros capítulos siga los menús desplegables para traducciones de acceso
El uso de traducción de Google, si las traducciones están equivocados, graciastambién me informó
Os doy las gracias de antemano.
Corinne
Allein zu seinbei der Schaffung dieserWebsiteohne die Hilfe vonjemand, wenn Sie vergessen, oder Fehlerauf meiner Seite, ich danke Ihnen bin, um per E-Mailzu informieren..wenn SieZeugnissemachen wollen,(natürlichanonym,die Zusatzinformationen, Fotos etc. kontaktieren Sie mich. ich würdeglücklich sein, siezu veröffentlichen.. . Darüber hinaus erleichtert das Lesen der jeweiligen Fremdsprache habe ich mehrere Schreibfarben verwendet folgenden Sprachen traduitent in der Homepage. In den weiteren Kapiteln folgen Sie die Dropdown-Menüs, um den Zugriff Übersetzungen
Verwendung von Google-Übersetzung, wenn dieÜbersetzungfalsch sind,dankeichauch darüber informiert,
Ichdanke Ihnen im Voraus.
Corinne
Будучив одиночестве всоздание этого сайтабез помощикого-либо, есливы видитезабыли, илиошибки намоем сайте, спасибомсообщитьпо электронной почте.. есливы хотите, чтобыпоказания, (конечно анонимныйпуть,дополнительная информация, фотографии и т.д., свяжитесь со мной.Я был бы счастлив, чтобы опубликовать их.. ,
Кроме того, для облегчения чтения каждого иностранного языка, я использовал несколько пишущих цвета следующих языках traduitent в домашней странице. В других главах следовать выпадающее меню для доступа переводов
Alleen zijn in het creëren van deze site zonder de hulp van iedereen, als je vergeten of fouten op mijn site, dank u ben op de hoogte via e-mail .. als je wilt getuigenissen maken (uiteraard anonieme manier , de aanvullende informatie, foto's, enz., contact met mij op. ik zou blij zijn om ze te publiceren ..
.
Bovendien, om te vergemakkelijken het lezen van elke vreemde taal, heb ik verschillende kleuren te schrijven volgende talen traduitent in de home page. In de andere hoofdstukken volgen de drop down menu's om toegang vertalingen. :
BLACK = french
BLUE = Engels
GROEN = Italiaans
SPAANSE = paars
DUITSE oranje =
BROWN Russische =
ROOD = Chinees (traditioneel)
DONKER GEEL = Nederlands
Met behulp van google vertaling, als de vertalingen zijn fout, dank u ook op de hoogte me
Ik dank u bij voorbaat.
Corinne
Eğer unuttuysanız, veya sitemde hatalar bkz eğer herkes yardımı olmadan bu siteyi oluştururken yalnız olmak, (siz ifadelerini yapmak istiyorsanız .. e-posta ile bilgilendirme m elbette anonim bir şekilde teşekkür , tamamlayıcı bilgiler, fotoğraflar, vs, bana ulaşın. Ben bunları yayınlamak için mutlu olurdu ..
.
Ayrıca, her yabancı dil okuma, ben ana sayfada diller traduitent aşağıdaki birkaç yazılı renkler kullandık kolaylaştırmak için. Diğer bölümlerde erişim çeviriler için aşağı açılır menülerle izleyin. :
SİYAH = fransız
MAVİ = İngilizce
YEŞİL = İtalyan
Mor = İSPANYOL
ALMAN turuncu =
KAHVE Rus =
KIRMIZI = Çince (Geleneksel)
KOYU SARI = Hollandalı
Ek diller:
Türk
Çeviriler yanlış ise google çeviri kullanarak, siz de beni bilgilendirdi teşekkür