
關於綜合徵
摘要
這正是格林綜合症? 定義
DR戈爾林RJ,外科醫生,牙醫和遺傳學家,它的路線
基底細胞痣綜合徵的遺傳特徵
A /一般
B /基因參與了基底細胞痣綜合徵
1.一般
雙重事件2.理論
3.傳導方式修補的/音速刺猬
演員通路修補/刺猬索尼克
修補工作/刺猬索尼克的傳導途徑
信號通路修補/刺猬索尼克
突變和位置的類型
詳細常規事件,基底細胞痣綜合徵
1 /活動DERMATOLOGICALS
A /痣和基底細胞癌一般
B /掌蹠角化過度通用
C /皮膚病事件
2 /骨架活動
A /腦鈣化信封
B /異常骷髏頭造型
C /肋骨畸形
D /脊椎畸形
手和腳的E /骨骼異常
F /其他骨骼表現
3 /眼睛活動
地球的A /先天畸形
B /腫瘤地球過程
4 /活動的神經系統
A /Méduloblastomes統計
B /其他神經系統表現
5 /其他普通活動
A /卵巢肌瘤
B /心臟肌瘤
C /性腺功能減退的男性:
6 /活動ODONTOSTOMATOLOGIQUES
A /牙源性的角化
B /其他事件odontostomatologiques
A /夾雜物或牙齒錯位
B /下頜前突
C /唇齶裂裂
D /感覺異常
E /的améloblastome
這正是格林綜合症? 定義
戈爾林綜合徵,也稱為基底細胞痣綜合徵(NBC)是一種遺傳性疾病,其特徵一套發育異常和易患各種癌症。
的患病率估計為1/57和1/256 000 000,具有比陽/陰1:1。 臨床表現包括眾多的基底細胞癌(BCC),頜牙源性角化囊腫,手掌和腳掌,骨骼異常角化,顱內鈣化異位和面部異形(大頭畸形,唇裂,腭存在而嚴重的眼部異常)。 知識赤字在5%的患者觀察到。 基底細胞癌(從具有皮膚潰瘍性板,其直徑為1至10毫米的彩色丘疹)通常位於面部,背部和胸部。 基底細胞癌的數量從幾個到幾千。 骨骼異常(影響肋骨,椎骨和顱骨的形狀)頻繁。 也可能會出現眼睛,泌尿生殖和心血管疾病。在患者戈爾林綜合徵,5-10%出現髓母細胞瘤,這是早期死亡的一個可能的原因。
綜合徵是由基因PTCH1突變引起的,被發送作為一個常染色體顯性遺傳完全外顯率和可變表現度。 臨床診斷依據特定的標準。 診斷是由基因突變的研究證實。 遺傳諮詢是強制性的。 產前診斷可以通過羊膜穿刺術或絨毛取樣獲得的胎兒細胞超聲和DNA分析。 其主要鑑別診斷包括Bazex綜合徵,多發性trichoépithélium和繆爾-托雷綜合症綜合症(參見這些術語)。 該支持需要多學科的方法。
角化通過手術切除除去。 在基底細胞癌的情況下,手術時指示病變的數目是有限的。 其它可能的治療是激光燒蝕,光動力療法,和局部化療。
應避免放射治療。 維生素A的類似物可以對戰的新CBC發展的預防作用。 預期壽命不顯著改變,但並發症可導致發病率顯著。 定期監測由多學科小組(皮膚科,神經科和odontologists)是必不可少的。 患者應避免過度暴露於紫外線。
出版商(S)的專家(S)
DR戈爾林RJ,CHRIRURGIEN-牙醫和遺傳學家,它的路線

羅伯特·詹姆斯·戈林生於1923年1月11日在Hudson,紐約和明尼阿波利斯,明尼蘇達州在83歲時死於淋巴瘤2006年8月29日。 二戰期間參軍時,他再攻讀牙科在華盛頓大學,他畢業於1947年,他通過獲得化學碩士在大學繼續他的教育州愛荷華州。 經過無數次的學術職務,在1956年,他獲得了位置,他在那裡,直到他去世教授和口腔病司司長明尼蘇達大學。
他的職業生涯轉了個彎,1940年,當戈林發現一本書的海倫Curth皮膚科醫生處理achantosis皮病。 這方面的經驗將有助於“syndromologiste”改造戈林。 該achantosis皮病是一種皮膚疾病,其特徵vegetating乳頭肥大和色素沉著主要分佈在腋下,頸部和生殖器,大腿區域的地方出現皮膚粗糙,增厚,平方。 這種皮膚病有特點是與胃腺癌的關聯惡性形式。它是一種皮膚病,影響身體其他系統。 這個發現迷住了戈林,然後問,如果某些全身性疾病會出現口腔表現。 戈林成為國際知名的許多症狀的口腔表現為他的工作。 在他的整個職業生涯中,羅伯特·戈林和他的同事們研究和命名了100症狀,並參加了隔離和負責其中一半的基因特徵。
戈林的名字已成為一個共同的名字,7不僅在遺傳學,而且在醫藥熟悉。 這促使皮膚科專家,遺傳學家和病理學家考慮戈林為其中之一。 羅伯特·戈林將是一個非常多產的作家。 他已經寫了600餘篇,60章的書,並合作撰寫或編輯20本書。 他們中的一些人甚至被翻譯成西班牙語,俄語和日語。 戈林的名字已成為從基底細胞痣綜合徵,通常也被稱為格林綜合症尤其是分不開的。
在1960年,描述了一種用於在第一時間的出現在整個身體,包括未曝光區域在太陽與肋畸形,骨病變和缺陷和眼瘤大量皮膚基底細胞癌的關聯。 他認為,這些異常現象作為一個單獨的綜合徵比今天在長期格林綜合症的一部分。 這將有助於理解和發現的致病基因,該基因PTCH的。
在他的職業生涯在明尼蘇達大學,廣博的知識使他放棄在病理學,皮膚科,兒科,產科,婦科和otorhino咽喉課程。 他也是國際牙科研究協會,口腔病理學的美國學院和顱面生物學國際協會主席。 由於他對科學,醫學與社會的巨大貢獻,羅伯特·戈林已經收到了許多獎項在他的職業生涯。 因此,羅伯特·戈林是一個訓練的牙醫,誰通過他的工作是能夠跨越醫學和遺傳學領域以及牙科。
基底細胞痣綜合徵的遺傳特徵
A.一般
龍戈爾林RJ,這種臨床實體,稱為術語基底細胞痣綜合徵之前,已經被這個問題說明,在其他的名字。 1894年雅裡施
簡要地描述了在從脊柱側凸的患者骨骼異常基底細胞癌的組合。 它並沒有提及其他的
該疾病的特徵。
同年,白報導多發性基底細胞癌,這繼續出現在他的生活病人的家族史。
1932年Nomland介紹先天性色素痣引起的基底細胞癌。
1939年Straith是第一個上頜囊腫多個皮膚基底細胞癌,在一個家庭的三個成員聯繫起來。
它會等到1951年兩個皮膚科醫生,賓克利和約翰遜形容這兩種皮膚癌和上頜竇囊腫和
大腦的變化。
這是在1960年在明尼阿波利斯的戈林,牙醫和戈爾茨,皮膚科執行此,他們認為一個綜合徵的臨床實體的第一個全面的說明。 他們會給他基底細胞痣綜合徵的上頜骨和雙歧肋骨多發囊腫的名稱。
此後,綜合徵可能由幾個名字如戈林綜合徵或戈爾林-戈爾茨戈爾林-戈爾茨phacomatosis,基底細胞痣綜合徵被指定...
基底細胞痣綜合徵的已知最早的案件被揭露在20世紀60年代,因為含牙囊腫,雙歧肋骨和格林綜合症等的柱頭上發現埃及收集兩個成年男性骨架骨架都靈大學的人類學研究所。 這些骨骼是從王朝時期發掘出來的Assyut在下埃及和日期。 王朝時期始於埃及,一旦分為兩個王國,北方和南方的統一,是公元前3000年左右。
基底細胞痣綜合徵是一種遺傳性遺傳性疾病與常染色體顯性遺傳。 然而,散發病例已經報導:在例的60%,沒有其它家族成員達到與這些情況下35-50%表示néomutations。 因此,沒有家族病史,不排除本病的診斷。 它是由PTCH基因的突變引起的,位於9q22.3-Q31,這是一種基因
腫瘤抑制基因。
的痣樣基底細胞具有高外顯率這與其中一個基因顯示其效果的頻率。據估計為97%。這意味著有97%
攜帶這種基因的個體患有該疾病的表現中的至少一個。
該疾病還具有可變表現度; 表現存在具有可變定量方面的基因的品質。該疾病本身表現在患者變量的許多症狀到另一個。有超過四十診斷標準。然而,每一個病人患有這種疾病並不提供所有的標準。診斷標準已被分為兩類,主要和次要的,這取決於
它們的發生頻率。它有可能使基底細胞痣綜合徵,當患者存在至少有兩個主要標準或同事的診斷
主要標準有兩種次要標準。
患病率五萬六千分之一與性別比例男/女3/1。據Ghailan,患病率六萬分之一和男性和女性都受到影響
相同的比例。這實現男女平等是很好的解釋由常染色體顯性遺傳綜合徵。
據沃爾夫,患病率範圍從六萬分之一到十二萬分之一和男女都同樣受到影響。此外綜合症發生在多種基團的
文化似乎不具有偏愛特定類型的皮膚。
基底細胞痣綜合徵仍然是目前,只有700例已文獻報導一種罕見的疾病。然而,牙醫是在第一位置做出診斷,因為頜面牙科表現有這種疾病的第一表現形式中。
B.基因參與了基底細胞痣綜合徵
1.一般
負責基底細胞痣綜合徵的基因PTCH基因,發現首次於1996年由兩個研究小組。他的發現是相當近期
導致這種疾病的所有遺傳機制沒有得到很好的科學界了解,一些元素仍不清楚。
位於染色體9的長臂和更精確地在9q22.3,該基因發現突變的患者戈爾林氏綜合徵,而且在例
散發性基底細胞癌。
此基因在胚胎發育中起重要作用和細胞增殖的控制。
在1982年的一篇文章,菲茨帕特里克提供了一個非常不同的理論目前的數據來解釋這種綜合徵的由來。當時的科學家他們的技術已經在事實上,未檢測到基因變異,我們今天所知道的。他說,多系統的異常無法解釋的酶缺陷的存在。的這種缺陷妊娠的前三個月期間,存在可能解釋的骨骼異常和鈣化信封大腦。
後來,在酶機制控制皮膚中的異常可能導致痣,基底細胞癌,掌蹠凹坑和牙源性角化的形成。然而這一理論是不夠的,因為它確實permetpas了解神經腫瘤如髓母細胞瘤的起源。
2.雙重事件理論
該研究的腫瘤在基底細胞痣綜合徵顯示出雜合性為9q的區域的損失的存在,這表明PTCH基因可能是腫瘤抑制基因。這個概念抑癌基因的,其原型是成視網膜細胞瘤基因(RB),根據該基因的兩個等位基因的在腫瘤從提出的努森模型中的損失(雜合13的損失)於1971年根據這兩個突變事件'的他的“理論,第一突變負責畸形綜合徵和易患腫瘤的發生和第二將導致腫瘤的出現。
根據科恩,使腫瘤抑制基因失活,這兩種現象是必需的。第一個是一個等位基因的轉移。第二個現象是另一個等位基因,被稱為雜合性(或LOH)的損失的損失。當兩個等位基因丟失,腫瘤發生。雜的損失證明為基底細胞癌,牙源性角化和髓母細胞瘤,這是三個主要特點戈爾林綜合徵。
因此,在家族性形式戈爾林綜合徵,第一突變可以存在於受孕,並在所有身體細胞被發現。這第一個突變事件,產生一個傾向開發基底細胞痣綜合徵的後代,在主導地位傳輸。這一事件仍然潛伏,只暴露,如果第二個事件發生在染色體
同源的相同基因座,從而導致外觀戈爾林綜合徵。在家族性病例,個人只需要一個單一的突變步驟(因為繼承了它的第一個遺傳物質)。通過對外觀格林綜合症的個人無家族史
需要兩個連續的突變事件在同一個體的發生。
3.傳導方式修補的/音速刺猬
該PTCH基因是果蠅基因修補的人類同源。這種基因是果蠅基因(或極性)的路徑中產生的分割
是參與胚胎發育和細胞增殖的控制轉導修補/音猬。它用作通過停止細胞分裂,在沒有配體和激活一旦連接是與配體的細胞週期調節。
格林綜合症導致大腦損傷,肋骨,椎骨,會員...修補的表達在這些組織中。
演員通路修補/音速刺猬:
儘管很大的進步已取得近年來在理解信號通路修補/刺猬的操作,很多
不確定因素仍然需要在今後加以澄清。
- 修補的苯硫脲或蛋白質的PTCH基因編碼的是一種糖蛋白由1447個氨基酸有十二個跨膜結構域,
所需的配位體SHH,寬細胞內循環,一個細胞內的氨基末端結構域和羧基末端胞內結構域的結合,以及兩個大胞外環。
發現突變在基底細胞痣綜合徵,而且在基底細胞癌,髓母細胞瘤和罕見的情況下。
大多數突變是導致截短的蛋白質的合成,無義突變。(拉孔布)基因修補1
-聲波刺猬蛋白(SHH)SHH編碼由位於7q36基因。此分子量約為分子量約為45kDa的蛋白裂解成兩個域:具有水解酶活性和25 kDa的的自催化賦予了羧基端結構域的20 kDa的鋅的的氨基末端結構域
膽固醇轉移酶活性。
膽固醇在這一信號通路的作用尚不完全清楚,但它可以在限制SHH分子和分佈的擴散發揮作用
空間效果。
- 最後,磨平蛋白(SMO),通過設在的7q31-32的Smoothened的基因編碼。
它是115 kDa的蛋白質,屬於偶聯至G蛋白蛇形受體家族(其示出同源性的家族的Wnt受體)787個氨基酸組成。後者有一個細胞外氨基末端結構域,羧基末端胞內結構域,以及兩個區域(第三胞內環和第七跨膜結構域)參與信號。
修補工作/刺猬索尼克的傳導途徑:
修補是一種受體SHH作用配體。在沒有SHH,SMO修補的,並形成無活性複合物。修補充當
負調節,SMO的抑製劑。
當SHH自帶結合到修補的,後者則是隨後滅活和SMO是自由地發送所述小區內的信號。
幾種機制已經提出來解釋SMO和信號轉導的活化:
- SMO複雜後者,因此離解的構象變化。SMO然後將釋放發送細胞內的信號。
-最近,一個第二假設提出了一種非化學計量的調控修補的。事實上,測試表明SHH結果diminutionde修補的綁定,並在細胞膜增加的GOS。之間的關係
出動和SMO將非化學計量的,即,將規範的修補16
SMO的變化,但不構成真正的複合物與它(會有兩者之間沒有物理交互)。
SMO被激活,並且可以執行信號轉導到細胞核。
通過GOS的精確信號轉導途徑尚不清楚,但已知的是最終的目標是由轉錄因子的GLI(肘interruptus的同源物或次果蠅)的活化來表示。在細胞未受到SHH GLItétramétrique形成與其他蛋白如沿海-2(cos2的)融合(富)和融合因子(SUFU)的抑制複雜。在這種形式下,GLI可裂解為75 kDa的的氨基末端片段,可以轉位到細胞核,抑制靶基因在後者。
在SHH配體的存在,所述複合物離解和全發揮其作用的GLI轉錄活化(發生在核心和允許靶基因的表達)。
靶基因是:
- 無翅(WG)WNT的家族的脊椎動物的編碼部件,
- Decapentaplegic(DPP)編碼骨形態發生蛋白(BMP)和的βTFG
- PTC編碼修補自己。
這些基因是正常胚胎發育和某些組織的分化所必需。
無翅基因編碼一個家庭參與,如分化,遷移,細胞增殖和極性發育過程的糖蛋白。該基因的表達失調負責發育異常和腫瘤形成。
該decapentaplegic基因起著對信息的胚胎外胚層的背腹分化傳輸一個小角色。
信號通路修補/刺猬索尼克
修補在發展中的作用:
一個Hedgehog信號通路在幹細胞的發育在各種組織中的重要作用。PTCH表示無論在
胚胎發育和成年人,這表明它仍然有在產後期的作用。表達不適當刺猬蛋白信號傳導途徑的任何組分實驗模型揭示顯著發育異常。這些異常與那些特徵戈爾林綜合徵之間的相似性暗示此信號傳導途徑的過度激活可在lacnaevomatose基底細胞發現發育異常和腫瘤的原因。
修補調節由SHH傳送的信號的傳輸,信號參與幾個embryonaux方法如腹側神經管的形成,腦,四肢或牙齒發育的零件...
突變和位置的類型
許多突變已報導由作者。實際上,基底細胞痣綜合徵不能歸因於單個突變。根據患者中,有義突變,錯義,倒位,缺失...它們可以分佈在PTCH基因的整個編碼序列。
大多數突變導致產生的這導致截短的蛋白質的合成,一個終止密碼子。
這些突變都集中為最寬細胞內循環,寬胞外環和PTCH基因的氨基末端區域上。
沒有相關性可在基因突變的位置和在不同的患者戈爾林綜合徵中觀察到的表型之間建立。 這種綜合徵的一個特徵是缺乏的基因型/表型的相關性。 為攜帶相同突變的個體的臨床症狀可以是非常可變的。 這表明的其他因素,例如環境或遺傳因素在該疾病的發展的影響的可能性。
詳細常規事件,基底細胞痣綜合徵
1 /活動DERMATOLOGICALS
A /痣和基底細胞癌一般:
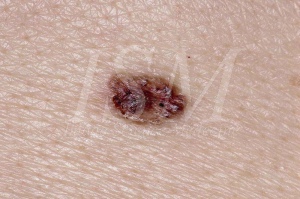
基底細胞痣是基底細胞痣綜合徵的主要標誌之一,使得在不存在這些痣; 一些醫生取出診斷標誌。 它們對於疾病的主要標準之一,在該牙源性角化的方式相同。 他們有惡性變的基底細胞癌的大進化潛力。
他們發現,在格林綜合症個案當中,超過90%。 據估計,200例基底細胞癌或多個達到戈爾林綜合徵。 更多基底細胞癌作出診斷,越早這一比例增加。 考慮到大約五分之一的患者為20歲前基底細胞癌選自基底細胞痣綜合徵患者。
發病年齡:
基底細胞癌,因為部分戈林綜合徵,出現在較早的年齡比正常人群。 他們可能是在出生時或兒童時期發展起來的。 大多數情況下,它們的發生開始於青春期和由40歲,90%的受這種疾病的患者已經開發的至少一個基底細胞癌。
號:基底細胞癌的數量是高度可變的。 雖然臨床病例已觀察到與單個癌,在大多數情況下,數為20和幾千之間。
地點:
這些腫瘤的位置可以在陽光暴露的區域(從而紫外線)比未曝光區域進行兩者。 眼瞼,鼻子,臉頰和額頭是受影響最大的網站,但頸部,軀幹及腋下也經常受到影響。 通過利弊,頭皮和四肢,大部分時間倖免。 臨床表現:基底細胞痣可以採用可變臨床方面。
三個方面一般描述:
- 外觀喚起傳染性軟疣contagium是一種傳染性皮膚狀況,並在一定條件下接種。 它是由病毒引起的,其特徵是亞光白色的小升高或玫瑰珍珠的大小和頂部也擴大了在肚臍凹陷。
- 沉著大結節的外觀。
- 外觀喚起古典基底細胞癌。 這是最常見的。 痣則顯示為一個小表面光滑透亮的腫瘤。 它的顏色是可變的,通常是正常的皮膚,有時呈粉紅色或灰色。 一致性是強得多比預期的外觀,但鹼沒有滲透。 的大小而變化,1至3毫米的直徑。如果任何這些病變的大小或覆蓋有外皮增加或開始出血成為,惡性轉化之嫌。 出現在基底細胞痣綜合徵內基底細胞癌,不能清楚地從那些出現在一個不受該綜合徵的患者區分開。 唯一的特徵元件戈爾林綜合徵是其在大的數字和在幼年和它們的位置,這不一定是在日光暴露的區域發生。 這些腫瘤的臨床表現是高度可變範圍從肉色丘疹棕色或潰瘍性斑塊。他們可以被誤認為是痣血管瘤或。 它們的大小而變化之間1和10毫米直徑。
組織學:
基底細胞痣的組織學外觀是上皮細胞的皮內擴散,形成明確的範圍,往往與外圍柵欄坐著。 下面的基質中含有豐富的成纖維細胞。 基底細胞癌患者戈林綜合徵的組織學表現是不是從那些非綜合徵患者中發現不同。 當痣變得基底細胞癌,人注意到更大,更清晰的細胞,有絲分裂的數量增加時,浸潤的炎症性字符。 它有一個纖維間質。 病變可成為papullaire或大步。 深穿透,潰瘍或入侵可能發生的淋巴細胞浸潤。 它們可被著色的質量或在外圍。
太陽的影響:
一項研究表明,患者的基底細胞痣綜合徵會呈現敏感性增加紫外線。 這是一個事實,即只有14%的患者格林綜合症在英格蘭西北部發展基底細胞癌,對澳大利亞47%的人支持。 它強調產前診斷(當達到一個父)的重要性,或儘早診斷(對於出現的情況下從頭),以限制經診斷UV曝光積極的。
另一項研究中,進行報告的婦女基底細胞癌和男子在總人口中86%以上的88%以上發生在暴露在陽光下對59%的婦女,基底細胞痣綜合徵的男性攜帶者中65%的身體區域。 他的結論是病變的解剖分佈表明,太陽是不是在基底細胞癌發展的一個重要因素。 他們在更大的數字出現在暴露部位,不過,暗示太陽會加劇基底細胞癌的發展,必須保護自己。 他沒有報告的基底細胞癌的數量和暴露於陽光下的小時總數之間的任何顯著的相關性。
只有約40%的非裔美國人格林綜合症的發展基底細胞癌對90%的白種人。 在105例戈爾林綜合徵的研究表明,僅38%的非洲裔美國人具有針對白種人的97%的一種或多種基底細胞癌。 此外,白種人更早期的發展基底細胞癌。 的確,白種人的大約50%發展其第一癌21.5年和90%,在35年的反抗20%和40%在相同的年齡在非裔美國人
該組的非裔美國人在最小頻率基底細胞癌,可能是由於對紫外線,由於他們的皮膚色素沉著更好的保護。
電離輻射的影響:
電離輻射也被牽連在這些腫瘤的發展。 事實上,放療治療的患者往往有在照射區眾多的基底細胞癌。 他們出現在振盪平均6個月,並針對與非患者格林綜合症平均21歲照射經過3年的期間。
應當指出的是,該疾病的早期標誌之一是髓母細胞瘤。 現在一般治療與放療。 在照射區域在顯影BCC的風險大大增加。此外,這些癌為更加積極,因此更難以治療。 由於這些原因,必須盡量減少放射治療和鼓勵手術治療。
進化:
基底細胞癌表現為部分戈林綜合徵顯得更積極,更難以治療。 他們可以由侵襲(轉移在腦和肺為主)或抗治療導致患者的死亡。 主要是青春期後發生惡變。 最侵襲性腫瘤最常影響眼瞼和鼻子和引起組織損傷,可能導致患者一個顯著毀容。
B /掌蹠角化過度通用:
他們還根據“坑”的條款得到滿足,孔或井掌蹠。 26頻率:黃蓉良在幼年時,它們的存在可能是基底細胞痣綜合徵的早期診斷的重要助劑,因為它們存在於大約65%至80%的病例。 他們會見了大致在所有年齡組相同的頻率。 掌在水中浸泡幾分鐘後角化手。
臨床表現:
它們對應於小孔是在角質層,這是部分地或完全不存在點狀缺陷。 很少超過1〜2毫米,直徑1毫米的深度,他們的背景可以是粉紅色,或者,如果污垢堆積在裡面的,黑暗的。 這些病變只在手和腳遇到並無症狀。 當臨床診斷是困難的,醫師可以浸泡幾分鐘的手和/或在溫水中的病人的腳。 該凹坑看起來更清楚呢。
組織學:
一個手掌坑組織學檢查顯示小前增生基底細胞。 雖然這方面的範圍內的蜂窩解體,未觀察到的前部和相鄰的基質之間的裂縫。 輕度血管增生和纖維化,觀察在相鄰的真皮層。 istologique
總之,掌蹠坑是一個特徵性表現格林綜合症。 他們是因為發生,其發生年齡提前了他們的高頻率,此綜合徵的診斷的重要輔助。 由於這些原因,它們中的疾病的主要的診斷標準,基底細胞癌和牙源性角化。
皮膚囊腫相對於基底細胞痣綜合徵的頻率是75%,這取決於該研究在35。 表皮樣囊腫通常發生在身體pilleuses領域,很少在非pilleuses領域,如手掌和腳掌。 在本體的端部這一典型位置將是基底細胞痣綜合徵的顯著的臨床表現。
此組織學描述非常類似於牙源性角化,基底細胞痣綜合徵的主要診斷標準。 這些皮膚角化組織學頜骨角化的親屬,可能是基底細胞痣綜合徵的典型商標。
總之,皮膚囊腫主要分佈在手腳的發現,一些有很強的組織學上的相似與牙源性角化囊腫,一定要算“臨床珍珠”,它可以帶領我們走向格林綜合症的診斷。
C /其他皮膚病表現
其他皮膚病表現被找到所引用的文獻中,但由於其非常低的發生頻率,都沒有明確說明。 因此,患者可有脂肪瘤,糧食粟丘疹,污點“拿鐵咖啡”或傳染性軟疣的鐘擺。 脂肪瘤位於在右側肩胛骨。 也注意到許多皮膚痣的存在。 該軟體動物鐘擺是纖維瘤和鬆弛的皮膚。 他們可能是平面的,電鍍,感覺鬱悶,或者相反,預測,甚至有蒂。 所述達米利亞是其特徵在於通過小升高形成皮膚的一種罕見的疾病亮半透明,針頭由上層的變性膠體(細胞在一種果凍的轉換)產生的尺寸真皮。
2 /骨架活動
A /腦鈣化信封
有可能在孩子的生理或病理顱內鈣化滿足。 前者是非常罕見的。 後者是更常見的,通常描述於相關的腫瘤(如髓母細胞瘤),神經皮膚綜合症,感染和神經變性疾病。 在一般人群中,顱內鈣化出現平均約為12年,而在患者格林綜合症,他們是從第一年的生活可見。 因此,它們可以用作早期指標為基底細胞痣綜合徵的診斷。 它們通常與假腦,這是硬腦膜成形兩個半球之間的中心分區的一部分。 他們的患者戈爾林綜合徵頻率證明相當重要的,因為65和根據作者85%之間振盪。 他們代表了最常見的放射性標誌。 在小腦幕的鈣化也遇到過,但更很少超過上錯了大腦。 小腦的帳篷是大腦和小腦之間硬腦膜的一部分。 也可以會見胼胝體發育不全,但這種異常依然罕見。
B /異常骷髏頭造型
患者基底細胞痣綜合徵具有顱骨的形狀的異常。 這些異常現象,雖然屬於疾病的次要標準,非常感興趣的診斷,因為它們是從出生可見。 在一些患者顱骨異常大突出與前額突出和頂葉,顳。 眉毛很突出。 該過遠(眼睛過度間隔)是常見的前突,有時報告。 鼻子的基地已折疊和展開。 一個鼻中隔偏曲,還可以觀察的105例格林綜合症的研究,大頭畸形,觀察更頻繁(佔50%),在這些患者比正常人群。 它是相同的hypertélorisme其存在於例的42%。
C /肋骨畸形
雖然它屬於疾病的次要標準,肋軟骨異常是存在的患者約42%,而其在群體中的頻率是很低的。 他們在很大程度上是當前和主要由代表: - bifidités - 發育不全 - 肋骨骨性癒合。 在骨性癒合是兩個相鄰的骨頭總焊縫。 另外,也可以觀察到肋的前或後端的異常的擴大。 第三,第四和第五肋最常受即使所有的肋可以實現這些缺陷。 像Kimonis一些作者決定該疾病的主要標準之一肋異常分類的原因有兩個。 第一種是它們的高頻率(42%)的患者相比,一般人群。 第二個是,這些異常可以很容易地在出生時檢測到的,因此代表在兒科人群的基底細胞痣綜合徵的主要診斷元件。 施加Kimonis的條件,可以在大量的病人中進行該疾病的早期診斷。 事實上,其它主要標準的疾病,如牙源性角化或多發性基底細胞癌,不存在在出生; 他們大多是兒童期或青春期出現。 臨床醫生應該非常關注這種疾病在兒童時期輕微的臨床症狀。 格林綜合症的存在,應尋求與生肋骨異常,心臟纖維瘤,或腭唇裂,多指畸形或大頭畸形兒。
D /脊椎畸形
據105例格林綜合症的研究中,被更頻繁地在該組的人影響了檢測脊柱側彎病例。 然而,該差異並不證明足夠大,是有意義的。 生病的人似乎有更嚴重的脊柱側彎較一般人群和最經常位於發育異常的網站,如半椎體融合或椎體。 這些發育異常的病例中有31%遇到過。 脊柱裂病例報告,但具有稍微不同的頻率比普通人群。 脊柱裂是一個缺陷,包括在一個或多個椎骨的默認脊柱骨化點,通過它們是疝氣中的或多或少的大腫瘤的形式的焊料後部軸的一條縫,該腦膜和有時與腦脊液的可變數量的骨髓。 頸胸列最受此缺陷影響的患者格林綜合症
手和腳的E /骨骼異常
多趾是不是一種非常常見畸形,因為它是在只有3%的情況下遇到的,但是,它是容易檢測及從出生本。 它的診斷容易。 患者戈爾林綜合徵還可以具有第四和/或第五掌骨的縮短。 這些畸形是該疾病的次要標準中,因為它們是罕見的。
F /其他骨骼表現
紫莖澤蘭畸形:患有基底細胞痣綜合徵11%有先天性畸形肩胛骨紫莖澤蘭或抬高。這種異常包括向上和向內的一個或兩個肩胛骨與變形,有時附著在移動向流離失所骨脊柱。此增加是由於骨的正常下降位置的胎兒期的失敗。
3 /眼睛活動
眼部受累的頻率仍不清楚。這是因為它們佔據小的空間中的說明戈爾林綜合徵,作為這種疾病最發表的研究治療主要標準。眼部異常不屬於提到的問題,不是一個真正的描述。
地球的A /先天畸形
遇到的主要眼缺陷是白內障,青光眼及缺損其頻率組合由戈爾林估計少於14%。
白內障是一種眼部疾病導致晶狀體混濁或它的膠囊。
青光眼是一種特徵為增加超過20毫米汞柱眼壓眼部情況。它是由於不適房水的正常流動。
缺損是由外圍切口,單個或多個透鏡的一個缺陷。
兩眼之間的過遠或過大的距離,也經常遇到,因為它是基底細胞痣綜合徵的典型面部特徵的一部分。
除了這些異常現象,也有可能找到斜視,先天性白內障,小眼球或色素性視網膜炎的案件。視網膜色素變性是視網膜,雙邊,家庭和遺傳性的退化過程。它出現在童年,其特徵是視敏度和視野狹窄的漸進性喪失,最終導致失明
B / 腫瘤地球過程
眼皮經常基底細胞痣的風險惡變的座位。該轉化可以通過建立一個遲鈍潰瘍的可表現(即,它不演變,並且不倒退)和侵入性或通過滲透深度與蓋切割。眼瞼基底細胞癌一般都比較積極,更難治療。此外,他們的治療可引起顯著面部切割為患者。
案瞼板腺囊腫也有報導。瞼板腺囊腫是從一個瞼板腺,皮脂腺跗軟骨慢性炎症小眼瞼腫瘤,構成眼瞼的框架。
4 /活動的神經系統
A /Méduloblastomes統計:
在一般人群中,髓母細胞瘤是在兒童中最常見的腦腫瘤及影響約20%的所有這些腫瘤。診斷的平均年齡為6年和3個月,男孩受到的影響比女孩更大的程度。這些腫瘤的傳統治療包括手術切除聯合放療和化療。存活率在5年時是60至80%。約小於5歲的患者與髓母細胞瘤的5%是由戈爾林綜合徵實現。這個速度下降到1%或2%的成年人的情況下。平均年齡髓母細胞瘤診斷的患者是早得多,因為它為2.1年。
由於這個原因,這是非常重要的考慮髓母細胞瘤中的第一歲的兒童的生活的存在下,在顯影基底細胞痣綜合徵的風險。
病人症狀有更好的存活率比非病人。
治療方法:
手術切除,放療和/或化療為輔,一般建議以消除髓母細胞瘤。在戈林綜合徵的情況下,開始這些腫瘤的年齡早; 金加放射療法被實踐,長期後遺症(如生長病症或內分泌功能障礙)越早風險是重要的。此外,在照射區域,顯影BCC或腦膜瘤的風險大大增加。這些基底細胞癌被證明是對治療更加積極和抵抗力。
由於這些原因,與放射治療感染症的人的治療,應盡可能避免。
髓母細胞瘤也滿足了42化療,這種治療方法應該更受歡迎,至少在年輕患者。
某些功能格林綜合症,目前在幼年時,如智力低下或大腦鐮鈣化,只能解釋為造成腫瘤的表現。此外,最經典的標誌性疾病,如皮膚損傷或上頜骨角化,只出現後。
由於這些原因,基底細胞痣綜合徵可能很難辨認在這個年齡儘管腫瘤的存在。
一些作者認為髓母細胞瘤應成為該疾病的主要標準之一,因為它是一部分,骨改建,最早的跡象。遺傳調查和嚴格的臨床檢查必須提供在尋找一個可能的高林綜合徵,所有患者的髓母細胞瘤3歲和他們的家人面前。
B /其他神經系統表現
腦膜瘤:
相關腦膜瘤基礎naevomatoses的只有10例已文獻報導,患者的年齡為18 64歲之間。沒有性的優勢。這些腫瘤出現在誘導由以前的髓母細胞瘤的治療輻射區域增長。
智力低下:
智力低下和學習困難的情況下也有報導患者約5%。
5 /其他普通活動
A /卵巢肌瘤:
受格林綜合症的女孩和婦女可出現卵巢肌瘤。肌瘤是最常見的卵巢的良性腫瘤,代表所有卵巢腫瘤的約20%。在1964年的初步報告,戈林和戈爾茨估計,卵巢肌瘤存在於生活與格林綜合症的女性75%。
一系列進行的研究最近發現的14至24%患病率。達到平均年齡30.6年,但可能從16-45年。但是請注意,某些腫瘤中發現患者年齡小於3.5年。出於這個原因,患者必須綜合徵13歲前接受他們的第一個超和定期做婦科護理。卵巢肌瘤生長緩慢和直徑6cm的平均尺寸,即使一些可能會高達30厘米的直徑。
據報導,在戈爾林綜合徵遇到卵巢肌瘤是更頻繁的雙邊和鈣化比在一般人群中。在大多數情況下,這些腫瘤完全無症狀。他們不妨礙生育和病人很少發生惡變。這些腫瘤的治療方法是手術。
B /心臟肌瘤:
心臟纖維瘤的病例報告與基底細胞痣綜合徵連接,但並不多見。它們是3至5%的患者具有心臟纖維瘤。他們代表只有5%的心臟腫瘤。他們出現在童年和在10歲以下的85%的情況下的兒童。他們最喜歡的地方是室間隔。這些腫瘤被快速發現,因為它們在生命的早期出現,並可能導致高達病人的早期死亡。
C /性腺功能減退的男性:
男子與格林綜合症可呈現很少性腺功能低下。性腺功能減退是受試者的生殖腺具有不足的內部分泌的條件。
6 /活動ODONTOSTOMATOLOGIQUES
A.牙源性角化
1.一般
在1956年描述的,第一次通過Philipsen,是一個牙源性角化,根據世界衛生組織或WHO,“良性單向或多細胞,骨內源性原點,具有特性層复層鱗狀上皮parakératinisé和潛在的攻擊行為和滲透。“ 世衛組織還建議將“腫瘤”一詞,因為這能更好地反映腫瘤(攻擊行為,主要的擴張,傾向復發...)的性質。術語原始囊腫長期以來被用作術語角化的同義詞,保持混亂。事實上,所有的原始囊腫是不是因為角化他們並不都角化。相反地,所有角化不在,只要它們不總是開發代替牙齒的必不可少囊腫。
術語表皮樣囊腫,同時,的代名詞。他們代表了約11%的頜骨囊腫一切的。他們排名第三,在神經根和濾泡囊腫後。它們出現三次更頻繁下頜骨比上頜骨。
該牙源性角化囊腫發生於各年齡組,雖然累積頻率為10次四十年間觀察到在正常人群中。通過利弊,患者格林綜合症,峰值頻率早,所以是人生的第一個十年。這些囊腫從而構成了疾病的早期症狀。這證實選擇牙醫在早期檢測基底細胞痣綜合徵的地方。他們是最偶然發現的時間段標準照相。無症狀的患者的百分比的變化取決於研究從34至50%。它們也可以通過症狀,如腫脹,疼痛最多外觀露出來的。但牙關緊閉,蜂窩組織炎,等等延遲萌出或牙齒錯位...
請注意,所有這些症狀都沒有具體的牙源性角化的。角化牙源性的年輕病人的存在或復發或多個角化外觀應提醒醫生和對格林綜合症的可能診斷。大約5%的角化的與此綜合徵有關。他們發現,在80%的患者戈爾林綜合徵當它們存在於只有5-7%的一般人群。有幾種理論來解釋牙源性角化囊腫的病因。
一些牙源性角化將開發從假牙。
事實上,以下牙胚的形成,牙帶溶解在兩個夾爪。在這一現象,小島和上皮細胞簇能堅持到結締組織。它將上皮這些殘基,其在活化時由未知因素是角化的形成的原因。根據另一方面,它們從釉質器官或口腔粘膜的上皮的基底層的星網狀發出。事實上,這等理論假定基底層的影響是角化囊腫形成的原因。一種觀點支持這一理論認為,這些囊腫復發的觀察,即使手術切除受影響的骨段。這假定復發的原點是這個骨骼之外,並且可能是在相鄰的軟組織。應當指出的是,這兩種理論並不是不相容的,因為這兩個牙板作為口腔粘膜負責外胚層組織。
2.特點
牙源性角化囊腫的病理診斷可以通過它的histolopathologique分析來完成。放射學檢查肯定能引導從業者,但在任何情況下做出明確的診斷。該考試由內容分析和囊性牆後是完整切除囊腫或採取它的一個片段,在他或她的marsupialisation引流後獲得。大多數作家列出的主要組織學特徵是那些Pinborg和漢森在1966年:
- 鱗狀上皮襯裡通常是非常細而均勻的厚度,很少或根本沒有味蕾,並具有較強的有絲分裂活動;
-一個定義良好的基底細胞層。了組成它的細胞是立方形或圓柱形的形狀和排列最經常在一個柵欄裝置;
- 薄薄的一層,從多刺細胞經常呈現的細胞內水腫的四到八個細胞層;
-角質是,通常,角化不全(核的存在下)。它發生在角化的83-97%。有時有可能滿足角化orthokeratosic樣(無核)或兩者對位和orthokeratosic。對於一些作家,如馬丁orthokeratosic那種應個體化,因為它會少些攻擊性和復發性的角化不全樣的少;
-角蛋白層通常是波浪形的。唯一的角化不是一個具體的標準,角化,因為其他牙源性囊腫可產生角蛋白。
- 纖維囊腫壁通常是薄,通常不發炎;
-常含上皮島或囊腫“女孩”隔離外圍殼薄結膜。囊腫的腸腔內容物有液體方面,面色蒼白或黃膩,更白,更濃。有角蛋白的存在,但在不同的量。在的情況下的炎症反應的在該內容膽固醇結晶(在光澤薄片形式存在),透明體,多核和細菌團塊被發現。另外,當囊腫壁發炎,上皮變厚並逐漸變成非角化上皮相當相似,牙源性炎症司空見慣囊腫。FNA也是牙源性角化囊腫的診斷的重要考驗。
事實上,這在角質FNA的出現是診斷輔助元素(但僅是不夠的,作出明確的診斷,因為其他的囊腫,如囊腫含牙,主根或有上皮表面可角化)。
然而,如上所述,炎症反應的存在下誘導的角化的parakératinisation的漸進性喪失,因此可以防止有時正確細胞學診斷。一個FNAB的轉診診斷角化其他元素是囊腫的蛋白質含量的估計。
當該內容被測量的至少4克每100毫升液體蛋白,診斷角化然後比較安全的。對於該測定,炎症的存在也有不好的影響。
事實上,它傾向於增加蛋白質含量,因此參與建立的假陰性。也有方法轉向手術前實現診斷。這些技術還沒有普遍的做法。首先是搜索KAC抗原(角化抗原)的囊液。這種存在證實了診斷。二是細胞角蛋白10的免疫組化表達的決心。
事實上,它是角化上皮的標記物。它是在約50%的牙源性角化的發現,而且在根性囊腫的10%,含牙囊腫的3%。然而角蛋白這不會是在見地作為這些病變的角化的表達的研究似乎減壓後失去的表達。
3.臨床和影像學特點
地點:
該角化牙源性座椅更經常在上頜骨下頜骨。事實上,它發生三次更經常在頜骨,特別是在角水平和支,隨後先在區域中的位置和第二臼齒和更前的位置。
在上頜骨角化囊腫的總部設在prémolomolaire地區和犬和極其前區早一點。
發病年齡:與格林綜合症相關的牙源性角化囊腫的發展,一般來說,人生的第一個十年期間和在20和30年達到其發病高峰期,不像與此綜合徵和角化相關後來被發現,第四個十年(平均年齡37歲)中。
因為這個早期的發展,它們構成了疾病的最初明顯的症狀之一,並確認所選擇的地方為牙科在這種綜合徵的診斷。
尺寸:
它們的體積更變,從幾毫米到一個半下頜骨(18)的洪水。因為它們常常後來發現,在一次例行X-射線檢查,這種情況並不少見,他們已經達到相當的體積。5厘米,平均長度。 (34)
性別比例:
在正常人群中男性往往比女性的2/1性別比例的影響。通過患者的基底細胞痣綜合徵利弊,兩性之間的差不存在,並且它甚至似乎婦女受影響稍微更經常比男性。(5)症狀:多數牙源性角化囊腫是沒有任何症狀。的確,34至50%是無症狀的,因此牙科或放射檢查程序中才發現。當症狀存在,它們最常發生由拱形或逐行骨腫脹的外觀可能是痛苦的。
炎症現象則並不少見,其特點是膿腫,蜂窩組織炎,甚至有時還伴有fistulisations一個破傷風的發生。這些現象之外,角化可以因為錯位,牙齒錯位咬合或延誤可能導致皮疹和這將挑戰從業者被發現。
應當指出的是,所有這些表現是不是在所有特定於牙源性角化,並將需要一個放射學檢查和特別是組織病理學以確認診斷。
增長:
其中一個牙源性角化囊腫的主要臨床特點是其強大的增長潛力。這甚至導致世衛組織認為他們為良性腫瘤的標準之一。各種理論已經提出來解釋這一特點:
-第一個解釋是在角化內部的膠原酶的酶的存在。她將負責這也解釋了這種強勁的增長潛力的膠原溶解活性;
- 另一種理論是骨吸收引起的前列腺素和電子網由囊腫分泌的存在;
-最後,囊液並增加有絲分裂功率囊腫壁的更大的滲透壓也可能是負責這一強勁的增長。延伸的這些囊腫的骨代松質骨,而不是作為其他孢囊朝皮質骨和骨膜區域橫向延伸範圍內。
當角化囊腫體積大,他們正在成型的骨骼和牙齒,皮質53(通常是外部的)的邊緣,然後變薄,有時穿孔。下頜骨和/或重大面部畸形骨折的病例報告,但卻很少見。高貴的元素,例如下牙槽管的神經血管束的,反過來,始終尊重,但可以變回(極少導致感覺異常)。
復發,復發的機制:
該牙源性角化囊腫也有非常高的復發率。這是他們的主要臨床特徵之一。這個速度是即使在患者基底細胞痣綜合徵和更高大約82%對在健康患者(33)的25-60%。有解釋的原因,再犯這種高速率三種機制:
- 囊腫由於非常薄的和易碎的囊腫壁或周圍的不完全切除,無論是由於皮質骨與囊腫粘附到鄰近的軟組織的穿孔;
- 牙源性角化從衛星囊腫,遺忘或手術後微囊殘存牙的發生;
-新的角化牙源性的在相鄰區域和其中的發展被解釋為復發,而它是一個從頭具有與原發病灶沒有連接形成。根據其他作者,復發率也受:
- 該尺寸,位置(拉莫斯),病變(尤其multiloculaire),使治療更加困難和手術的形態
- 手術技術的選擇(保守或激進的)大多數復發被發現在5-7年的第一個支持。
然而,病例報告治療超過10年。復發病例25%位於9歲以上。的牙源性角化治療54一個基本方面,在長期監測(10年似乎理想能夠在早期診斷幾乎所有復發)。牙源性角化囊腫中的某些功能帶來了世衛組織重新分類為腫瘤或良性腫瘤,而不是囊腫: -第一行為:這些囊腫是當地極具破壞性,浸潤性,具有較高的復發率; -然後其組織病理學:研究顯示,基底層芽中的結締組織。世界衛生組織還指出,有絲分裂數字經常發現在超基底層; 上皮角化有絲分裂指數增加相對於另一個鉗口囊腫(但類似於成釉細胞瘤); -要完成的基因:該基因PTCH是位於染色體9q22.3-Q31一種腫瘤抑制基因。該基因負責提供基底細胞痣綜合徵,而且負責的孤立牙源性角化的形成。通常PTCH編碼跨膜蛋白,其形成與癌基因的SMO(平滑)為配體SHH(音猬)的信號接收器。SHH受體激活此時,有不同的組織和細胞增殖的生長信號的抑制。它也參與脂質代謝的調節。通過利弊,如果一個人失去了工作PTCH,我們將有一個細胞增殖的影響(因此發展囊腫和腫瘤)。
標準放射性方面:
很常無症狀,多數在角化一個標準的放射學檢查被發現。這種考試在任何情況下做出明確的診斷。它允許診斷的取向只能由組織病理學分析來確認。牙源性角化的最常見的X射線的外觀是單(通常)或polygéodes透亮,圓形或橢圓形,並與薄內部分區。
在上頜骨,覆蓋竇或鼻腔可以使這些特徵的識別變得更加困難。當角化非常廣泛,細化和皮質骨變形是可能的。它可以發展任何半側頜骨; 髁總是尊重。
後者還可以換一個unevolved鄰牙(含稅),一個生牙或odontome並導致位移,或者很少,對位於接觸其他牙齒的牙根吸收。周邊高尚元素(神經...)沒有被破壞,而是由囊腫送回。主要的鑑別影像學診斷與出現造釉細胞瘤,囊腫含牙那麼一些。
4.進化
鱗狀細胞癌角化牙源性惡變的病例非常罕見。只有12個案件已文獻報導至今。當有多個復發,病變的一個非常積極的行為,以及兩者的保守治療並積極的治療失敗的這種可能性,應考慮。它往往是在老年患者中觀察到。案例成釉細胞轉化的報導,但也很少見
。5. 鑑別診斷
牙源性角化囊腫的鑑別診斷必須與其他三個主要囊腫做:
- 與成釉細胞瘤:
這囊腫是最常見的男性和具有其峰值發生率在第四和第五數十年。80%的成釉細胞瘤的位於下頜骨和特別是在角的水平和支。上頜本地化是罕見的。
從臨床角度看,造釉細胞瘤的特點是無限生長和高復發率。這些病變通常有足夠大的容量。一般無症狀,將只能通過影像學檢查偶然發現。否則,它可以由感染發作或由運動和牙齒異常進化來揭示。
從一個放射性點,造釉細胞瘤大多一個polygéodique方面的“肥皂泡”,定期邊界和內部分區。Geodes是很好劃定不同大小的,圓形或橢圓形。高貴的元素被壓抑,不被破壞。有這麼多的相似之處與角化,使鑑別診斷的唯一標準的臨床和影像學表現是不可能的。鑑別診斷將因此主要是由組織學檢查也由CT掃描。
- 用含牙囊腫:
此囊腫在男性滿足更頻繁具有其峰值發生率,在第二和第三十年。它主要涉及第三下臼齒和上犬齒和下第二前磨牙。
較自由的症狀時,含牙囊腫附著一個阻生牙的頸部和圍繞冠。他的發現是最常見的在一次例行X線檢查或由不明原因的牙齒移動的動機。
該X線表現是,一個良好的劃定骨溶解,monogéodique和發展圍繞花圈或冠阻生牙的,完全建成。病變可能會變得非常大,侵犯下頜骨升支。這是更難以察覺放射學插入它在阻生牙的頸部,因此,微分一個角化。
另外,有可能觀察靠近病灶齒的根吸收; 這對於牙源性角化非常罕見,是這兩個實體的主要特色的放射性標誌。
- 隨著卵泡囊腫:
這種囊腫是各種含牙囊腫,但它與牙齒接觸的毛囊發展; 也就是說,牙齒不完全形成的。它的發現可以通過用恆齒的延遲噴發動機X光片的方式進行; 任一過程中的感染性發作(通常在臼齒區域)。其他特徵是相同的含牙囊腫。
- 隨著一個平庸的炎性囊腫或牙科radiculo:
這些病變是最常見的頜骨。它們優先地發生在上頜前或下頜骨的水平分支男性。他們的發病高峰是第三和第四之間的幾十年。同樣病變之前看到的,根尖囊腫通常無症狀和放射性日常發現。不過,可以透露的特點是鼓鼓的,腫脹,流膿一個齲齒,壞死或不完全處理的旁邊感染發作。這些囊腫緩慢生長,並且可以,在不存在的臨床症狀,展開相當。
他們的放射線照相外觀是一個monogéode的,圓形或橢圓形,與羞愧和由邊框骨縮合(其可在炎症發作消失)包圍的牙齒的頂端連接。他們可能會,如果顯著的大小,相鄰根的原因牙根吸收。 59
鑑別診斷也可以與其他囊腫土地,如牙源性纖維瘤,牙源性的粘液瘤,孤囊腫,骨血管瘤或動脈瘤囊腫。60 B.
B /其他事件odontostomatologiques
其他牙科odonto-事件也遇到了基底細胞痣綜合徵,但有較大的差距。事實上,它們並不總是存在,並且當它們是,它是一個具有頻率比牙源性角化它們是,本身的疾病的主要標準之一要低得多。他們往往直接關係到角化的發展和可能代表的第一次調用標誌之一,突出他們(並因此導致基底細胞痣綜合徵的診斷)
A /夾雜物或牙齒錯位
他們主要關注的恆牙。它有可能觀察到的初級齒的持久性時的最終牙齒由囊腫,這防止其噴發保留。研究已證實,取決於囊腫的時間發展,後果牙科水平是不同的。實際上,當角化開發時根構造,觀察到形變在根部諸如曲率或彎曲刺刀。
如果它的發展並置早至牙科元件形成,可以使根部的撕裂。通過利弊,如果囊腫牙齒萌出後形成的,他可能是負責旅遊和牙齒錯位,但在任何情況下,可以不負責的牙根吸收或紙漿屈辱。
B /下頜前突
病例的大約25%。它可以是一個真實的前突或連接於brachymaxillie一個pseudoprognathisme,或兩者的組合。61他們的支持將通過正頜外科手術和正畸。在年輕的患者高林綜合徵下頜前突(15)
C /唇齶裂裂
唇裂和/或腭裂被發現在大約5-7%基底細胞痣綜合徵的病例。通過Lambrecht的進行的文獻回顧顯示,受高林綜合徵719人,顏面部有61插槽,8.5患者%。這些人可分為根據時隙是否是陰唇,肺泡和腭(49例)或僅裂兩組(13例)
這些畸形的頻率不是一般人群和患者的基底細胞痣綜合徵之間的相同
。的確,在一般人群中,唇縫,肺泡和上顎的發病率是,僅1/800和1/2500針對分別腭裂1 / 14.7的患者1 / 55.3格林綜合症。這種異常可能是一個戈爾林綜合徵的新生的存在的第一指示中的一個。
ð。/感覺異常
它們也與角化的發展並且當囊腫達到顯著大小,如果其壓縮或壓制一個神經元,並導致感覺異常發生。它也可以是除去大囊腫與鄰近的神經邊界的術後並發症。
E /的améloblastome
在成釉細胞瘤的患者格林綜合症很少遇到幾起案件已文獻報導。伊斯拉米表示68歲的病人被格林綜合症,並具有成釉細胞瘤實現的情況下。
雖然受這種疾病影響,幾乎所有的患者,首先出現的症狀出現在幼年時,一般35歲之前,患者的首次臨床跡象開始出現得更晚。
事實上,它提出了它的第一個基底細胞癌在50歲和多個角化距離只有68年。有趣的元素,我們伊斯拉米指出,3的4例成釉細胞瘤記錄在英國文學相關的基底細胞痣綜合徵,也提出了他們的第一個症狀的年齡比大多數基底細胞痣綜合徵的病例。
平均年齡為這些情況下47.6年。這一發現表明,患者基底細胞痣綜合徵在以後年齡更處於發展成釉細胞瘤的風險。
由上可見,牙源性角化囊腫和成釉細胞瘤是,兩者具有很高的復發率和骨破壞的巨大潛力良性腫瘤。
然而,造釉細胞瘤是更持久,更浸潤,需要一種更為激進的手術切除。
SOURCES
